A pipeline for Whole Exome Sequencing (WES) variants identification in mathced tumor-normal samples. It runs into a Docker container, ready to be downloaded and used on any operating system supported by Docker.
All the steps of the pipeline and their dependencies are controlled by SCons so that in case of any stop, like killing by error or even shutting down the computer, it will automatically resume the analysis from the last run process.
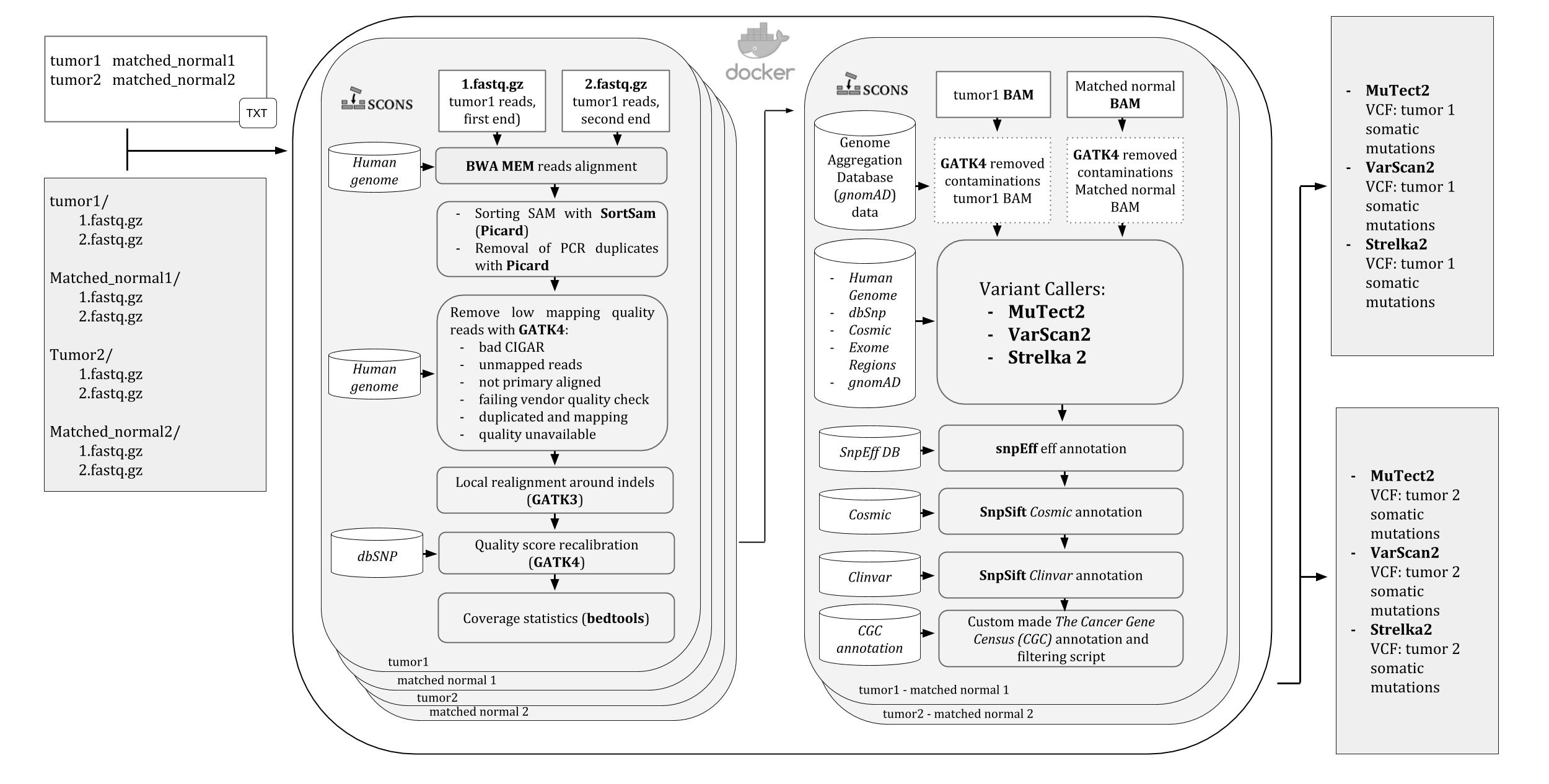
Three variant calling softwares are used by the pipeline: Mutect2 , VarScan2, and Strelka2 and the user is allowed to choose which to use and change their default settings.
It runs on both genomes versions: GRCh37 and GRCh38 (beta version).
A pdf manual for iWhale is available here
An image of the iWhale pipeline diagram is available here
{kind=link}
Binatti A, Bresolin S, Bortoluzzi S, Coppe A. iWhale: A Computational Pipeline Based on Docker and SCons for Detection and Annotation of Somatic Variants in Cancer WES. Briefings in Bioinformics. 2020 May 20:bbaa065. doi: 10.1093/bib/bbaa065.
Download iWhale from Docker Hub:
docker pull alexcoppe/iwhale
The working directory must contain the following elements:
- two directories for each sample: one for tumor and the other one for control samples. Each directory must include two fastq files
- a text file called tumor_control_samples.txt
- a python file called configuration.py
The working directory must not contain other directories except the ones of the samples indicated above
Each sample must be in its own directory containing the two paired-end gz-compressed fastq files. The files must be called:
- 1.fastq.gz
- 2.fastq.gz
The file tumor_control_samples.txt is a simple text file organized by two columns separated by tab: in the first column there are tumor directories names and in the second one the matched control directories names
tumor_sample1 control_sample1
tumor_sample2 control_sample2
...
The configuration.py file is essential. It is used to set parameters of the tools used by iWhale. All the possible parameters that you can set are gathered and explained in this file: configuration.py.
The configuration.py file is set for GRCh38 genome analyses.
For GRCh37 you can use a configuration.py file with only e parameter set (exomeRegions): the exome regions in bed format, which is a very important parameter (the only one you must have to specify for GRCh37 version of the genome). To do it use the exomeRegions parameter which default value is:
exomeRegions = "exome_regions.bed"
You have to change it to the exome interval used by your sequencing experiment (SureSelect_Human_All_Exon_V5 in the example below).
exomeRegions = "SureSelect_Human_All_Exon_V5.bed"
iWhale also needs the gziped version (made by bgzip) of the file and the .tbi index done using with these parameters (you need tabix and bgzip commands):
bgzip -c SureSelect_Human_All_Exon_V5.bed > SureSelect_Human_All_Exon_V5.bed.gz
tabix -p bed -S 3 SureSelect_Human_All_Exon_V5.bed.gz
Every MuTect2 and VarScan2 parameters can be set by using the configuration.py file. For example:
mutect2Parameters = "--normal-lod 2.5 --native-pair-hmm-threads 6"
varScanParameters = "--tumor-purity 2 --p-value 0.05"
Annotation data, except COSMIC files, can be downloaded from compgen.bio.unipd.it.
For the GRCh37 genome this are the links:
- Tar.gz version: http://compgen.bio.unipd.it/downloads/annotations.tar.gz
- Zip version: http://compgen.bio.unipd.it/downloads/annotations.zip
For the GRCh38 genome this are the links:
- Tar.gz version: http://compgen.bio.unipd.it/downloads/annotations_GRCh38.tar.gz
Decompression takes a while. The version of used databases are listed below ("Databases currently used" section).
COSMIC files, which are free only for academic researchers, can be downloaded from https://cancer.sanger.ac.uk/cosmic/download after # and login. The needed files are:
- CosmicCodingMuts.vcf.gz
- cancer_gene_census.csv
- CosmicCodingMuts.vcf.gz.tbi
**Download the right version (GRCh38 or GRCh37) based on the data you are using The CosmicCodingMuts.vcf.gz.tbi should be made by the user. See the Testing the Docker image section to see how to make it.
The docker's run command is used to mention that we want to create an instance of an image. The image is then called a container. This is the command to launch an iWhale container:
docker run --rm -it --name iwhalexp -v $(pwd):/working -v /home/user/databases:/annotations alexcoppe/iwhale
- --rm removes the container (it is optional)
- -it used for interactive processes (like a shell)
- --name used to name the container. If you do not assign a container name with the --name option, then the daemon generates a random string name for you
- -v used to share the two folders that iWhale needs: the working directory (used in the example the current directory by $(pwd)) and the folder including the databases files (in the example, /home/user/databases)
- iwhale is the name of the docker image to be run
- iwhalexp is the name of the iWhale docker container while iwhale is the name of the image
You can download a small random sample (107M) in tgz format or as a zip file for a fast testing of iWhale. It contains 1 simulated small tumor (tumor_sample) and 1 control sample (control_sample), a configuration.py file and a tumor_control_samples.txt files ready. Then do the following steps:
-
Install Docker Community Edition (CE) in your computer. Instructions and downloading links are here: macOS, Windows and Ubuntu
-
Download and install the iWhale images with this command:
docker pull alexcoppe/iwhale
-
Download and decompress annotations.tar.gz or annotations.zip for GRCh37 or annotations_GRCh38.tar.gz for GRCh38
-
Download the CosmicCodingMuts.vcf.gz and cancer_gene_census.csv files from COSMIC version 37 or version 38 of the genome using your credentials. You need to create a .tbi file from the CosmicCodingMuts.vcf.gz with these commands (needed the tabix and bgzip softwares from Samtools):
gunzip CosmicCodingMuts.vcf.gz
bgzip -c CosmicCodingMuts.vcf > CosmicCodingMuts.vcf.gz
tabix -p vcf CosmicCodingMuts.vcf.gz
- Finally launch iWhale from the iwhale_example directory with a command similar to the following one. Just remember that the path indicated in the command, /path_to_user_annotations_directory, should be changed to the real path were you decompressed the annotations.tar.gz file, for example /home/user/annotations:
Version GRCh37:
docker run --rm -it --name iwhalexp -v $(pwd):/working -v /path_to_user_annotations_directory_v37:/annotations alexcoppe/iwhale
Version GRCh38:
Download the configuration.py file which is required to run the GRCh38, put it in the analyses directory and run:
docker run --rm -it --name iwhalexp -v $(pwd):/working -v /path_to_user_annotations_directory_38:/annotations alexcoppe/iwhale
Paths in Windows should be written differently. For example:
docker run --rm -it --name iwhalexp -v c:/Users:/working -v c:/Users/annotations:/annotations alexcoppe/iwhale
Read Get started with Docker for Windows for a tutorial on using Docker on Windows.
In case of iWhale accidentally stopped, you have to re-launch the same Docker container that was running before stopping. To do this use the following command:
docker start -a iwhalexp
Obviously the name iwhalexp is the same of used by the docker run used in the "Launching iWhale" above section
The final results are obtained by merging the VCFs produced by each chosen variant caller (MuTect2, VarScan2,Strelka2) and are located in the Combined_VCFs_by_sample directory included in the VCF directory. For each matched-samples pair, the called snps and indels are put into two different vcf files. In particular their name will be tumor_control_merged_typeofvariants.vcf. The index files of VCFs are also present in the directory. Here is an example of results:
[root@iwhale Combined_VCFs_by_sample] ls -l
-rw-r--r-- 1 root root 46578 Mar 15 15:24 5_6_merged_indels.vcf
-rw-r--r-- 1 root root 1477 Mar 15 15:24 5_6_merged_indels.vcf.idx
-rw-r--r-- 1 root root 54011 Mar 15 15:24 5_6_merged_snps.vcf
-rw-r--r-- 1 root root 3354 Mar 15 15:24 5_6_merged_snps.vcf.idx
-rw-r--r-- 1 root root 46578 Mar 15 15:25 7_8_merged_indels.vcf
-rw-r--r-- 1 root root 1477 Mar 15 15:25 7_8_merged_indels.vcf.idx
-rw-r--r-- 1 root root 54011 Mar 15 15:25 7_8_merged_snps.vcf
-rw-r--r-- 1 root root 3354 Mar 15 15:25 7_8_merged_snps.vcf.idx
The VCFs produced by each variant caller are present in the upper directory called VCF. The Variants directory contains the intermediate files produced by the pipeline divided by software and pair of matched-samples directories.
| Program | Description | Version |
|---|---|---|
| Burrows-Wheeler Aligner (BWA) | BWA is a software package for mapping low-divergent sequences against a large reference genome. | 0.7.17 |
| Picard | A set of command line tools for manipulating high-throughput sequencing (HTS) data and formats. | 2.17.11 |
| GATK4 | GATK4 is the first and only open-source software package that covers all major variant classes for both germline and cancer genome analysis. | 4.0.6.0 |
| GATK3 | A variety of tools with a primary focus on variant discovery and genotyping. | 3.8-1 |
| Strelka2 | Strelka2 is a fast and accurate small variant caller optimized for analysis of germline variation in small cohorts and somatic variation in tumor/normal sample pairs. | 2.9.2 |
| VarScan | VarScan is a platform-independent software tool developed at the Genome Institute at Washington University to detect variants in NGS data. | 2.4.2 |
| SnpEff | Genomic variant annotations and functional effect prediction toolbox. | 4_3t |
| bedtools | Collectively, the bedtools utilities are a swiss-army knife of tools for a wide-range of genomics analysis tasks. | 2.17.0 |
iWhale uses databases and sequences indicated in the table below.
| Sequences or Databases | Description | Version |
|---|---|---|
| Human genome database | Feb. 2009 assembly of the human genome (hg19,GRCh37 Genome Reference Consortium Human Reference 37 | hg19 or GRCh37 |
| dbSNP | dbSNP contains human single nucleotide variations, microsatellites, and small-scale insertions and deletions | All_20180418.vcf.gz |
| gnomAD | The Genome Aggregation Database (gnomAD), developed by an international coalition of investigators, with the goal of aggregating and harmonizing both exome and genome sequencing data from a wide variety of large-scale sequencing projects | 2018-05-22 |
| SnpEff GRCh37 | SnpEff annotation for the human genome reference genome GRCh37) | GRCh37 |
| ClinVar | ClinVar aggregates information about genomic variation and its relationship to human health | 20190311 |
| COSMIC | COSMIC, the Catalogue Of Somatic Mutations In Cancer, is the world's largest and most comprehensive resource for exploring the impact of somatic mutations in human cancer | v89 |
| Sequences or Databases | Description | Version |
|---|---|---|
| Human genome database | GRCh38 version of the human genome | hg19 or GRCh37 |
| dbSNP | dbSNP contains human single nucleotide variations, microsatellites, and small-scale insertions and deletions | All_20180418.vcf.gz |
| gnomAD | The Genome Aggregation Database (gnomAD), developed by an international coalition of investigators, with the goal of aggregating and harmonizing both exome and genome sequencing data from a wide variety of large-scale sequencing projects | 2.1 |
| SnpEff GRCh38 | SnpEff annotation for the human genome reference genome GRCh38) | GGRCh38.86 |
| ClinVar | ClinVar aggregates information about genomic variation and its relationship to human health | 20200316 |
| COSMIC | COSMIC, the Catalogue Of Somatic Mutations In Cancer, is the world's largest and most comprehensive resource for exploring the impact of somatic mutations in human cancer | v90 |
All commands are launch from the directory containing the downloaded data. Many of the command take a LOT OF TIME to conclude.
The reference genome can be downloaded from ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/ by using these commands:
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.1.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.2.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.3.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.4.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.5.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.6.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.7.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.8.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.9.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.10.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.11.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.12.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.13.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.14.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.15.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.16.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.17.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.18.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.19.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.20.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.21.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.22.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.X.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.Y.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.chromosome.MT.fa.gz
The reference genome of Epstein-Barr virus (EBV) is available in FASTA format from https://www.ncbi.nlm.nih.gov/nuccore/AJ507799.2?report=fasta
Compress EBV reference genome into a .gz file
gzip EBV_reference_genome.fa
Join all chromosomes into one big FASTA file with all human genome in it from FASTA headers
zcat Homo_sapiens.GRCh38.dna_sm.chromosome.1.fa.gz Homo_sapiens.GRCh38.dna_sm.chromosome.2.fa.gz \
Homo_sapiens.GRCh38.dna_sm.chromosome.3.fa.gz Homo_sapiens.GRCh38.dna_sm.chromosome.4.fa.gz \
Homo_sapiens.GRCh38.dna_sm.chromosome.5.fa.gz Homo_sapiens.GRCh38.dna_sm.chromosome.6.fa.gz \
Homo_sapiens.GRCh38.dna_sm.chromosome.7.fa.gz Homo_sapiens.GRCh38.dna_sm.chromosome.8.fa.gz \
Homo_sapiens.GRCh38.dna_sm.chromosome.9.fa.gz Homo_sapiens.GRCh38.dna_sm.chromosome.10.fa.gz \
Homo_sapiens.GRCh38.dna_sm.chromosome.11.fa.gz Homo_sapiens.GRCh38.dna_sm.chromosome.12.fa.gz \
Homo_sapiens.GRCh38.dna_sm.chromosome.13.fa.gz Homo_sapiens.GRCh38.dna_sm.chromosome.14.fa.gz \
Homo_sapiens.GRCh38.dna_sm.chromosome.15.fa.gz Homo_sapiens.GRCh38.dna_sm.chromosome.16.fa.gz \
Homo_sapiens.GRCh38.dna_sm.chromosome.17.fa.gz Homo_sapiens.GRCh38.dna_sm.chromosome.18.fa.gz \
Homo_sapiens.GRCh38.dna_sm.chromosome.19.fa.gz Homo_sapiens.GRCh38.dna_sm.chromosome.20.fa.gz \
Homo_sapiens.GRCh38.dna_sm.chromosome.21.fa.gz Homo_sapiens.GRCh38.dna_sm.chromosome.22.fa.gz \
Homo_sapiens.GRCh38.dna_sm.chromosome.X.fa.gz Homo_sapiens.GRCh38.dna_sm.chromosome.Y.fa.gz \
Homo_sapiens.GRCh38.dna_sm.chromosome.MT.fa.gz EBV_reference_genome.fa.gz > reference.fa
It produces many files:
- reference.fa.amb
- reference.fa.ann
- reference.fa.bwt
- reference.fa.pac
- reference.fa.sa
This step takes a lot of time:
bwa index reference.fa
The produced files is:
- reference.dict
java -jar ~/local/picard.jar CreateSequenceDictionary R=reference.fa O=reference.dict
The produced files is:
- reference.fa.fai
samtools faidx reference.fa
wget ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b151_GRCh38p7/VCF/All_20180418.vcf.gz
gunzip All_20180418.vcf.gz
cat All_20180418.vcf | sed 's/>chr/>/' > All_20180418_nochr.vcf
The new version of dbSNP included AA field in INFO with special characters not allowed in VCF files
java -jar SnpSift.jar rmInfo All_20180418_nochr.vcf AA > All_20180418_noAA.vcf
mv All_20180418_noAA.vcf All_20180418.vcf
Need to install bgzip and tabix in your computer. Produced file:
- All_20180418.vcf.gz
- All_20180418.vcf.gz.tbi
This step takes a lot of time
bgzip -c All_20180418.vcf > All_20180418.vcf.gz
tabix -fp vcf All_20180418.vcf.gz
Download gnomAD (version 2.0.2) data.
wget http://compgen.bio.unipd.it/downloads/gnomad.exomes.r2.0.2.sites.vcf.gz
wget http://compgen.bio.unipd.it/downloads/gnomad.exomes.r2.0.2.sites.vcf.gz.tbi
The gnomad.exomes.r2.0.2.sites.vcf.gz is compressed by bgzip and indexed by tabix.
Download of ClinVar data from https://ftp.ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh38/. Obtained files:
- clinvar.vcf.gz
- clinvar.vcf.gz.tbi
Their date portion could be different, if so, remember to put the right filename in the configuration.py file:
clinvar = "clinvar.vcf.gz"
COSMIC files, which are free only for academic researchers, can be downloaded from https://cancer.sanger.ac.uk/cosmic/download after # and login. The needed files are:
- CosmicCodingMuts.vcf.gz
- cancer_gene_census.csv
Making the .tbi file:
gunzip CosmicCodingMuts.vcf.gz
java -jar SnpSift.jar rmInfo CosmicCodingMuts.vcf AA > CosmicCodingMuts_noAA.vcf
mv CosmicCodingMuts_noAA.vcf CosmicCodingMuts.vcf
bgzip -c CosmicCodingMuts.vcf > CosmicCodingMuts.vcf.gz
tabix -p vcf CosmicCodingMuts.vcf.gz