{kind=link}
HBV Integration Finder (HBVIF) pipeline.
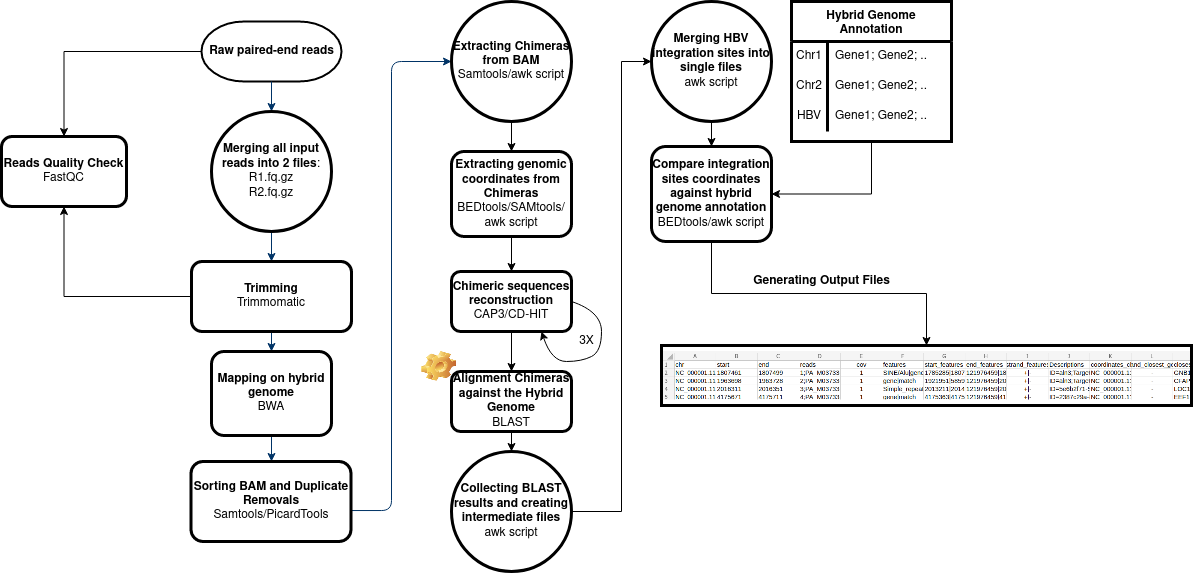
This bioinformatics pipeline was designed to directly extract the HBV-DNA integrated sequences from genomic breakpoints of the host genome (Human Genome). This pipeline takes as input directly the raw reads, performs the trimming (customizable/skippable), and processes them generating several tab-separated files that can be easily inspected.
Dependencies
Please be sure that the following software are properly installed on your system. The pipeline was designed and tested only on Ubuntu OS (18.04 LTS, 20.04 LTS) machines, using the following versions of softwares:
- fastqc (v0.11.8)
- trimmomatic (v.0.39)
- bwa (v 0.7.17-r1188)
- samtools (v. 1.9)
- picard tools (MarkDuplicates v. 2.20.1-SNAPSHOT)
- bedtools (v.2.28.0)
- cap3 (VersionDate: 02/10/15)
- cd-hit (v.4.8.1)
- blast (v.2.9.0-2)
The pipeline should work also with the most recent version of the above-mentioned tools, but keep in mind that this was not tested.
IMPORTANT!!! After you copy the repository using 'git clone https://github.com/DomeJoyce/HBVIF.git'
move into the genome folder and download the Human Genome from ncbi website, following the instruction present in the "prepare_genomes.txt"
This tool was developed by Domenico Giosa (dgiosa@unime.it) at the University of Messina, in collaboration with Sequentia Biotech SL (https://www.sequentiabiotech.com/).