-
Notifications
You must be signed in to change notification settings - Fork 13
Commit
This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository.
- Loading branch information
Showing
161 changed files
with
597,204 additions
and
188 deletions.
There are no files selected for viewing
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,203 @@ | ||
| mmCIF/ | ||
| .vscode/ | ||
| ckpt/ | ||
| multirun/ | ||
| wandb/ | ||
| *.pdb | ||
| *.csv | ||
| *.fa | ||
| *pdbs.jsonl | ||
|
|
||
| inference_outputs/ | ||
| # Byte-compiled / optimized / DLL files | ||
| __pycache__/ | ||
| *.py[cod] | ||
| *$py.class | ||
|
|
||
| # C extensions | ||
| *.so | ||
|
|
||
| # Distribution / packaging | ||
| .Python | ||
| build/ | ||
| develop-eggs/ | ||
| dist/ | ||
| downloads/ | ||
| eggs/ | ||
| .eggs/ | ||
| lib/ | ||
| lib64/ | ||
| parts/ | ||
| sdist/ | ||
| var/ | ||
| wheels/ | ||
| share/python-wheels/ | ||
| *.egg-info/ | ||
| .installed.cfg | ||
| *.egg | ||
| MANIFEST | ||
|
|
||
| # PyInstaller | ||
| # Usually these files are written by a python script from a template | ||
| # before PyInstaller builds the exe, so as to inject date/other infos into it. | ||
| *.manifest | ||
| *.spec | ||
|
|
||
| # Installer logs | ||
| pip-log.txt | ||
| pip-delete-this-directory.txt | ||
|
|
||
| # Unit test / coverage reports | ||
| htmlcov/ | ||
| .tox/ | ||
| .nox/ | ||
| .coverage | ||
| .coverage.* | ||
| .cache | ||
| nosetests.xml | ||
| coverage.xml | ||
| *.cover | ||
| *.py,cover | ||
| .hypothesis/ | ||
| .pytest_cache/ | ||
| cover/ | ||
|
|
||
| # Translations | ||
| *.mo | ||
| *.pot | ||
|
|
||
| # Django stuff: | ||
| *.log | ||
| local_settings.py | ||
| db.sqlite3 | ||
| db.sqlite3-journal | ||
|
|
||
| # Flask stuff: | ||
| instance/ | ||
| .webassets-cache | ||
|
|
||
| # Scrapy stuff: | ||
| .scrapy | ||
|
|
||
| # Sphinx documentation | ||
| docs/_build/ | ||
|
|
||
| # PyBuilder | ||
| .pybuilder/ | ||
| target/ | ||
|
|
||
| # Jupyter Notebook | ||
| .ipynb_checkpoints | ||
|
|
||
| # IPython | ||
| profile_default/ | ||
| ipython_config.py | ||
|
|
||
| # pyenv | ||
| # For a library or package, you might want to ignore these files since the code is | ||
| # intended to run in multiple environments; otherwise, check them in: | ||
| # .python-version | ||
|
|
||
| # pipenv | ||
| # According to pypa/pipenv#598, it is recommended to include Pipfile.lock in version control. | ||
| # However, in case of collaboration, if having platform-specific dependencies or dependencies | ||
| # having no cross-platform support, pipenv may install dependencies that don't work, or not | ||
| # install all needed dependencies. | ||
| #Pipfile.lock | ||
|
|
||
| # poetry | ||
| # Similar to Pipfile.lock, it is generally recommended to include poetry.lock in version control. | ||
| # This is especially recommended for binary packages to ensure reproducibility, and is more | ||
| # commonly ignored for libraries. | ||
| # https://python-poetry.org/docs/basic-usage/#commit-your-poetrylock-file-to-version-control | ||
| #poetry.lock | ||
|
|
||
| # pdm | ||
| # Similar to Pipfile.lock, it is generally recommended to include pdm.lock in version control. | ||
| #pdm.lock | ||
| # pdm stores project-wide configurations in .pdm.toml, but it is recommended to not include it | ||
| # in version control. | ||
| # https://pdm.fming.dev/#use-with-ide | ||
| .pdm.toml | ||
|
|
||
| # PEP 582; used by e.g. github.com/David-OConnor/pyflow and github.com/pdm-project/pdm | ||
| __pypackages__/ | ||
|
|
||
| # Celery stuff | ||
| celerybeat-schedule | ||
| celerybeat.pid | ||
|
|
||
| # SageMath parsed files | ||
| *.sage.py | ||
|
|
||
| # Environments | ||
| .env | ||
| .venv | ||
| env/ | ||
| venv/ | ||
| ENV/ | ||
| env.bak/ | ||
| venv.bak/ | ||
|
|
||
| # Spyder project settings | ||
| .spyderproject | ||
| .spyproject | ||
|
|
||
| # Rope project settings | ||
| .ropeproject | ||
|
|
||
| # mkdocs documentation | ||
| /site | ||
|
|
||
| # mypy | ||
| .mypy_cache/ | ||
| .dmypy.json | ||
| dmypy.json | ||
|
|
||
| # Pyre type checker | ||
| .pyre/ | ||
|
|
||
| # pytype static type analyzer | ||
| .pytype/ | ||
|
|
||
| # Cython debug symbols | ||
| cython_debug/ | ||
|
|
||
| # PyCharm | ||
| # JetBrains specific template is maintained in a separate JetBrains.gitignore that can | ||
| # be found at https://github.com/github/gitignore/blob/main/Global/JetBrains.gitignore | ||
| # and can be added to the global gitignore or merged into this file. For a more nuclear | ||
| # option (not recommended) you can uncomment the following to ignore the entire idea folder. | ||
| #.idea/ | ||
| data/data/ | ||
| outputs/ | ||
|
|
||
| #PDB processed dataset | ||
| data/processed_pdb/ | ||
| data/processed_pdb_openfold/ | ||
|
|
||
| #PDBBind dataset | ||
| DiffusionProteinLigand/data/PDBBind* | ||
|
|
||
| #pynb for debug code | ||
| pynb/ | ||
|
|
||
| #slurm_output for record | ||
| slurm_output/ | ||
| slurm-*.out | ||
|
|
||
| # chekpoint, and logs. | ||
| ckpt/ | ||
| *.pth | ||
| *.sw? | ||
| wandb/ | ||
| results/ | ||
|
|
||
|
|
||
| # keep track of the results example | ||
| !results_example/ | ||
| !results_example/length_50/* | ||
| !results_example/length_50/sample_0/* | ||
| !results_example/length_50/sample_0/self_consistency/* | ||
| !results_example/length_50/sample_0/self_consistency/esmf/* | ||
| !results_example/length_50/sample_0/self_consistency/seqs/* |
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,21 @@ | ||
| MIT License | ||
|
|
||
| Copyright (c) 2022 Justas Dauparas | ||
|
|
||
| Permission is hereby granted, free of charge, to any person obtaining a copy | ||
| of this software and associated documentation files (the "Software"), to deal | ||
| in the Software without restriction, including without limitation the rights | ||
| to use, copy, modify, merge, publish, distribute, sublicense, and/or sell | ||
| copies of the Software, and to permit persons to whom the Software is | ||
| furnished to do so, subject to the following conditions: | ||
|
|
||
| The above copyright notice and this permission notice shall be included in all | ||
| copies or substantial portions of the Software. | ||
|
|
||
| THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR | ||
| IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, | ||
| FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE | ||
| AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER | ||
| LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, | ||
| OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE | ||
| SOFTWARE. |
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,106 @@ | ||
| # ProteinMPNN | ||
| 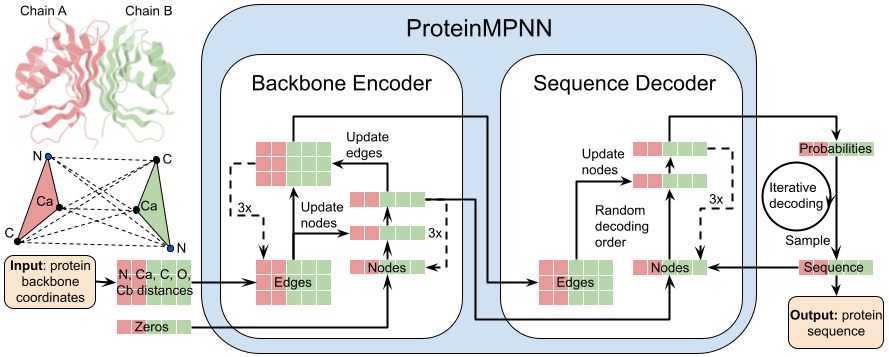 | ||
| Read [ProteinMPNN paper](https://www.biorxiv.org/content/10.1101/2022.06.03.494563v1). | ||
|
|
||
| To run ProteinMPNN clone this github repo and install Python>=3.0, PyTorch, Numpy. | ||
|
|
||
| Full protein backbone models: `vanilla_model_weights/v_48_002.pt, v_48_010.pt, v_48_020.pt, v_48_030.pt`. | ||
|
|
||
| CA only models: `ca_model_weights/v_48_002.pt, v_48_010.pt, v_48_020.pt`. Enable flag `--ca_only` to use these models. | ||
|
|
||
| Helper scripts: `helper_scripts` - helper functions to parse PDBs, assign which chains to design, which residues to fix, adding AA bias, tying residues etc. | ||
|
|
||
| Code organization: | ||
| * `protein_mpnn_run.py` - the main script to initialialize and run the model. | ||
| * `protein_mpnn_utils.py` - utility functions for the main script. | ||
| * `examples/` - simple code examples. | ||
| * `inputs/` - input PDB files for examples | ||
| * `outputs/` - outputs from examples | ||
| * `colab_notebooks/` - Google Colab examples | ||
| * `training/` - code and data to retrain the model | ||
| ----------------------------------------------------------------------------------------------------- | ||
| Input flags for `protein_mpnn_run.py`: | ||
| ``` | ||
| argparser.add_argument("--ca_only", action="store_true", default=False, help="Parse CA-only structures and use CA-only models (default: false)") | ||
| argparser.add_argument("--path_to_model_weights", type=str, default="", help="Path to model weights folder;") | ||
| argparser.add_argument("--model_name", type=str, default="v_48_020", help="ProteinMPNN model name: v_48_002, v_48_010, v_48_020, v_48_030; v_48_010=version with 48 edges 0.10A noise") | ||
| argparser.add_argument("--seed", type=int, default=0, help="If set to 0 then a random seed will be picked;") | ||
| argparser.add_argument("--save_score", type=int, default=0, help="0 for False, 1 for True; save score=-log_prob to npy files") | ||
| argparser.add_argument("--save_probs", type=int, default=0, help="0 for False, 1 for True; save MPNN predicted probabilites per position") | ||
| argparser.add_argument("--score_only", type=int, default=0, help="0 for False, 1 for True; score input backbone-sequence pairs") | ||
| argparser.add_argument("--conditional_probs_only", type=int, default=0, help="0 for False, 1 for True; output conditional probabilities p(s_i given the rest of the sequence and backbone)") | ||
| argparser.add_argument("--conditional_probs_only_backbone", type=int, default=0, help="0 for False, 1 for True; if true output conditional probabilities p(s_i given backbone)") | ||
| argparser.add_argument("--unconditional_probs_only", type=int, default=0, help="0 for False, 1 for True; output unconditional probabilities p(s_i given backbone) in one forward pass") | ||
| argparser.add_argument("--backbone_noise", type=float, default=0.00, help="Standard deviation of Gaussian noise to add to backbone atoms") | ||
| argparser.add_argument("--num_seq_per_target", type=int, default=1, help="Number of sequences to generate per target") | ||
| argparser.add_argument("--batch_size", type=int, default=1, help="Batch size; can set higher for titan, quadro GPUs, reduce this if running out of GPU memory") | ||
| argparser.add_argument("--max_length", type=int, default=200000, help="Max sequence length") | ||
| argparser.add_argument("--sampling_temp", type=str, default="0.1", help="A string of temperatures, 0.2 0.25 0.5. Sampling temperature for amino acids. Suggested values 0.1, 0.15, 0.2, 0.25, 0.3. Higher values will lead to more diversity.") | ||
| argparser.add_argument("--out_folder", type=str, help="Path to a folder to output sequences, e.g. /home/out/") | ||
| argparser.add_argument("--pdb_path", type=str, default='', help="Path to a single PDB to be designed") | ||
| argparser.add_argument("--pdb_path_chains", type=str, default='', help="Define which chains need to be designed for a single PDB ") | ||
| argparser.add_argument("--jsonl_path", type=str, help="Path to a folder with parsed pdb into jsonl") | ||
| argparser.add_argument("--chain_id_jsonl",type=str, default='', help="Path to a dictionary specifying which chains need to be designed and which ones are fixed, if not specied all chains will be designed.") | ||
| argparser.add_argument("--fixed_positions_jsonl", type=str, default='', help="Path to a dictionary with fixed positions") | ||
| argparser.add_argument("--omit_AAs", type=list, default='X', help="Specify which amino acids should be omitted in the generated sequence, e.g. 'AC' would omit alanine and cystine.") | ||
| argparser.add_argument("--bias_AA_jsonl", type=str, default='', help="Path to a dictionary which specifies AA composion bias if neededi, e.g. {A: -1.1, F: 0.7} would make A less likely and F more likely.") | ||
| argparser.add_argument("--bias_by_res_jsonl", default='', help="Path to dictionary with per position bias.") | ||
| argparser.add_argument("--omit_AA_jsonl", type=str, default='', help="Path to a dictionary which specifies which amino acids need to be omited from design at specific chain indices") | ||
| argparser.add_argument("--pssm_jsonl", type=str, default='', help="Path to a dictionary with pssm") | ||
| argparser.add_argument("--pssm_multi", type=float, default=0.0, help="A value between [0.0, 1.0], 0.0 means do not use pssm, 1.0 ignore MPNN predictions") | ||
| argparser.add_argument("--pssm_threshold", type=float, default=0.0, help="A value between -inf + inf to restric per position AAs") | ||
| argparser.add_argument("--pssm_log_odds_flag", type=int, default=0, help="0 for False, 1 for True") | ||
| argparser.add_argument("--pssm_bias_flag", type=int, default=0, help="0 for False, 1 for True") | ||
| argparser.add_argument("--tied_positions_jsonl", type=str, default='', help="Path to a dictionary with tied positions") | ||
| ``` | ||
| ----------------------------------------------------------------------------------------------------- | ||
| For example to make a conda environment to run ProteinMPNN: | ||
| * `conda create --name mlfold` - this creates conda environment called `mlfold` | ||
| * `source activate mlfold` - this activate environment | ||
| * `conda install pytorch torchvision torchaudio cudatoolkit=11.3 -c pytorch` - install pytorch following steps from https://pytorch.org/ | ||
| ----------------------------------------------------------------------------------------------------- | ||
| These are provided `examples/`: | ||
| * `submit_example_1.sh` - simple monomer example | ||
| * `submit_example_2.sh` - simple multi-chain example | ||
| * `submit_example_3.sh` - directly from the .pdb path | ||
| * `submit_example_3_score_only.sh` - return score only (model's uncertainty) | ||
| * `submit_example_4.sh` - fix some residue positions | ||
| * `submit_example_4_non_fixed.sh` - specify which positions to design | ||
| * `submit_example_5.sh` - tie some positions together (symmetry) | ||
| * `submit_example_6.sh` - homooligomer example | ||
| * `submit_example_7.sh` - return sequence unconditional probabilities (PSSM like) | ||
| * `submit_example_8.sh` - add amino acid bias | ||
| ----------------------------------------------------------------------------------------------------- | ||
| Output example: | ||
| ``` | ||
| >3HTN, score=1.1705, global_score=1.2045, fixed_chains=['B'], designed_chains=['A', 'C'], model_name=v_48_020, git_hash=015ff820b9b5741ead6ba6795258f35a9c15e94b, seed=37 | ||
| NMYSYKKIGNKYIVSINNHTEIVKALNAFCKEKGILSGSINGIGAIGELTLRFFNPKTKAYDDKTFREQMEISNLTGNISSMNEQVYLHLHITVGRSDYSALAGHLLSAIQNGAGEFVVEDYSERISRTYNPDLGLNIYDFER/NMYSYKKIGNKYIVSINNHTEIVKALNAFCKEKGILSGSINGIGAIGELTLRFFNPKTKAYDDKTFREQMEISNLTGNISSMNEQVYLHLHITVGRSDYSALAGHLLSAIQNGAGEFVVEDYSERISRTYNPDLGLNIYDFER | ||
| >T=0.1, sample=1, score=0.7291, global_score=0.9330, seq_recovery=0.5736 | ||
| NMYSYKKIGNKYIVSINNHTEIVKALKKFCEEKNIKSGSVNGIGSIGSVTLKFYNLETKEEELKTFNANFEISNLTGFISMHDNKVFLDLHITIGDENFSALAGHLVSAVVNGTCELIVEDFNELVSTKYNEELGLWLLDFEK/NMYSYKKIGNKYIVSINNHTDIVTAIKKFCEDKKIKSGTINGIGQVKEVTLEFRNFETGEKEEKTFKKQFTISNLTGFISTKDGKVFLDLHITFGDENFSALAGHLISAIVDGKCELIIEDYNEEINVKYNEELGLYLLDFNK | ||
| >T=0.1, sample=2, score=0.7414, global_score=0.9355, seq_recovery=0.6075 | ||
| NMYKYKKIGNKYIVSINNHTEIVKAIKEFCKEKNIKSGTINGIGQVGKVTLRFYNPETKEYTEKTFNDNFEISNLTGFISTYKNEVFLHLHITFGKSDFSALAGHLLSAIVNGICELIVEDFKENLSMKYDEKTGLYLLDFEK/NMYKYKKIGNKYVVSINNHTEIVEALKAFCEDKKIKSGTVNGIGQVSKVTLKFFNIETKESKEKTFNKNFEISNLTGFISEINGEVFLHLHITIGDENFSALAGHLLSAVVNGEAILIVEDYKEKVNRKYNEELGLNLLDFNL | ||
| ``` | ||
| * `score` - average over residues that were designed negative log probability of sampled amino acids | ||
| * `global score` - average over all residues in all chains negative log probability of sampled/fixed amino acids | ||
| * `fixed_chains` - chains that were not designed (fixed) | ||
| * `designed_chains` - chains that were redesigned | ||
| * `model_name/CA_model_name` - model name that was used to generate results, e.g. `v_48_020` | ||
| * `git_hash` - github version that was used to generate outputs | ||
| * `seed` - random seed | ||
| * `T=0.1` - temperature equal to 0.1 was used to sample sequences | ||
| * `sample` - sequence sample number 1, 2, 3...etc | ||
| ----------------------------------------------------------------------------------------------------- | ||
| ``` | ||
| @article{dauparas2022robust, | ||
| title={Robust deep learning--based protein sequence design using ProteinMPNN}, | ||
| author={Dauparas, Justas and Anishchenko, Ivan and Bennett, Nathaniel and Bai, Hua and Ragotte, Robert J and Milles, Lukas F and Wicky, Basile IM and Courbet, Alexis and de Haas, Rob J and Bethel, Neville and others}, | ||
| journal={Science}, | ||
| volume={378}, | ||
| number={6615}, | ||
| pages={49--56}, | ||
| year={2022}, | ||
| publisher={American Association for the Advancement of Science} | ||
| } | ||
| ``` | ||
| ----------------------------------------------------------------------------------------------------- |
Binary file not shown.
Binary file not shown.
Binary file not shown.
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1 @@ | ||
| <a href="https://colab.research.google.com/github/dauparas/ProteinMPNN/blob/main/colab_notebooks/quickdemo.ipynb" target="_parent"><img src="https://colab.research.google.com/assets/colab-badge.svg" alt="Open In Colab"/></a> |
Oops, something went wrong.