
An efficient tool to remove sequencing duplications and eliminate sequencing errors by generating consensus reads.
- What's gencore
- Download, compile and install
- Why to use gencore
- Understand the output
- How it works
- Command examples
- UMI format
- All options
- Read/cite gencore paper
gencore is a tool for fast and powerful deduplication for paired-end next-generation sequencing (NGS) data. It is much faster and uses much less memory than Picard and other tools. It generates very informative reports in both HTML and JSON formats. It's based on an algorithm for generating consensus reads, and that's why it's named gencore.
Basically, gencore groups the reads derived from the same original DNA template, merges them by generating a consensus read, which contains much less errors than the original reads.
gencore supports the data with unique molecular identifiers (UMI). If your FASTQ data has UMI integrated, you can use fastp to shift the UMI to read query names, and use gencore to generate consensus reads.
This tool can eliminate the errors introduced by library preparation and sequencing processes, and consenquently reduce the false positives for downstream variant calling. This tool can also be used to remove duplicated reads. Since it generates consensus reads from duplicated reads, it outputs much cleaner data than conventional duplication remover. Due to these advantages, it is especially useful for processing ultra-deep sequencing data for cancer samples.
gencore accepts a sorted BAM/SAM with its corresponding reference fasta as input, and outputs an unsorted BAM/SAM.
- Sample HTML report: http://opengene.org/gencore/gencore.html
- Sample JSON report: http://opengene.org/gencore/gencore.json
- BAM file for testing: http://opengene.org/gencore/input.sorted.bam
- BED file for testing: http://opengene.org/gencore/test.bed
- Reference genome file: ftp://ftp.ncbi.nlm.nih.gov/sra/reports/Assembly/GRCh37-HG19_Broad_variant/Homo_sapiens_assembly19.fasta
- Command for testing:
gencore -i input.sorted.bam -o output.bam -r Homo_sapiens_assembly19.fasta -b test.bed --coverage_sampling=50000- After the processing is finished, check the
gencore.htmlandgencore.jsonin the working directory. The option--coverage_sampling=50000is to change the default setting(coverage_sampling=10000)to generate smaller report files by reducing the coverage sampling rate.
The simplest way
gencore -i input.sorted.bam -o output.bam -r hg19.fastaWith a BED file to specify the capturing regions
gencore -i input.sorted.bam -o output.bam -r hg19.fasta -b test.bedOnly output the fragment with >=2 supporting reads (useful for aggressive denoising)
gencore -i input.sorted.bam -o output.bam -r hg19.fasta -b test.bed -s 2conda install -c bioconda gencoreThis binary is only for Linux systems: http://opengene.org/gencore/gencore
# this binary was compiled on CentOS, and tested on CentOS/Ubuntu
wget http://opengene.org/gencore/gencore
chmod a+x ./gencore# step 1: download and compile htslib from: https://github.com/samtools/htslib
# step 2: get gencore source (you can also use browser to download from master or releases)
git clone https://github.com/OpenGene/gencore.git
# step 3: build
cd gencore
make
# step 4: install
sudo make installAs described above, gencore can eliminate the errors introduced by library preparation and sequencing processes, and consenquently it can greatly reduce the false positives for downstream variant calling. Let me show your an example.
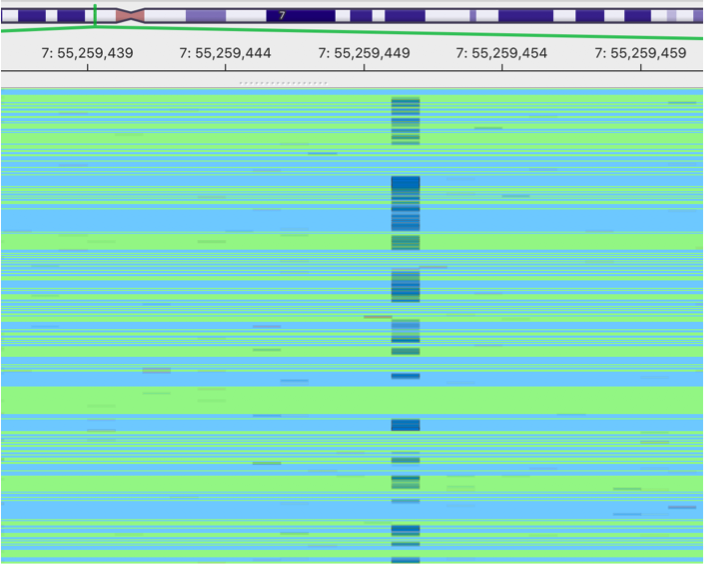
This is an image showing a pileup of the original BAM. A lot of sequencing errors can be observed.

This is the image showing the result of gencore processed BAM. It becomes much cleaner. Cheers!
gencore also performs some quality control when processing deduplication and generating consensus reads. Basically it reports mapping rate, duplication rate, mismatch rate and some statisticical results. Especially, gencore reports the coverate statistics of input BAM file in genome scale, and in capturing regions (if a BED file is specified).
gencore reports the results both in HTML format and JSON format for manually checking and downstream analysis. See the examples of interactive HTML report and JSON reports.


gencore outputs following files:
- the processed BAM. In this BAM, each consensus read will have a tag
FR, which meansforward read number of this consensus read. If the read is a duplex consensus read, it will also has a tagRR, which meansreverse read number of this consensus read. Downstream tools can read theFRandRRtags for further processing or variant calling. In following example, the first read is a single-stranded consensus sequence (only has aFRtag), and the second read is a duplex consensus sequence (has bothFRandRRtags):
A00250:28:H2HC3DSX2:1:1117:3242:5321:UMI_GCT_CTA 161 chr12 25377992 60 143M = 25378431 582
GCAATAATTTTTGTCAGAAAAATGCATTAAATGAATAACAGAATTTCTGTTGGCTTTCTGGGTATTGTCTTTCTTTAATGAGACCTTTCTCCAGAAATAAACACATCCTCAAAAAAATTCTGCCAAAGTAAAATTCTTCAAAT FFFFFFFFFFFFFFFFFFF,FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF NM:i:1 MD:Z:34G108 AS:i:138 XS:i:21 RG:Z:cfdna FR:i:2
A00250:28:H2HC3DSX2:1:2316:10547:25989:UMI_AAC_AGA 161 chr12 25377993 60 143M = 25378462 612
CAATAATTTTTGTCAGAAAAATGCATTAAATGAATAACAGAATTTCTGTTGGCTTTCTGGGTATTGTCTTTCTTTAATGAGACCTTTCTCCAGAAATAAACACATCCTCAAAAAAATTCTGCCAAAGTAAAATTCTTCAAATA FFFFF:FFFFFFFFFFFFFFFFFFFFF:FF:FFFFFFFFFF,FFFFFFFFFFFF,:FFFFFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFF:FFF,!FF:F:F:F,FFF,F:FFFF,,:F,FFFF:FF:,:FF:F,:, NM:i:1 MD:Z:33G67A41 AS:i:133 XS:i:21 RG:Z:cfdna FR:i:1 RR:i:5
- the JSON report. A json file contains lots of statistical informations.
- the HTML report. A html file visualizes the information of the JSON.
- the plain text output.
important steps:
- clusters the reads by their mapping positions and UMIs (if UMIs are applicable).
- for each cluster, compares its supporting reads number (the number of reads/pairs for this DNA fragment) with the threshold specified by
supporting_reads. If it passes, start to generate a consensus read for it. - if the reads are paired, finds the overlapped region of each pair, and scores the bases in the overlapped regions according their concordance and base quality.
- for each base position at this cluster, computes the total scores of each different nucleotide (A/T/C/G/N).
- if there exists a major nucleotide with good quality, use this nucleotide for this position; otherwise, check the reference nucleotide from reference genome (if reference is specified).
- when checking the reference, if there exists one or more reads are concordant with reference genome with high quality, or all reads at this positions are with low quality, use the reference nucleotide for this position.
gencore uses 3 different thresholds, and they can be specified by the commandline options:
| Quality threshold | Default Score | CMD option |
|---|---|---|
| High Quality | 30 (Q30) | --high_qual |
| Moderate Quality | 20 (Q20) | --moderate_qual |
| Low Quality | 15 (Q15) | --low_qual |
gencore assigns a score to each base in a read of a read cluster, the score means the confidence of this base. The score is given by following rules:
| in overlapped region? | matched with its pair? | condition? | score for this base |
|---|---|---|---|
| NO | N/A | HIGH_QUAL <= this_qual | 8 |
| NO | N/A | MODERATE_QUAL <= this_qual < HIGH_QUAL | 6 |
| NO | N/A | LOW_QUAL <= this_qual < MODERATE_QUAL | 4 |
| NO | N/A | this_qual < LOW_QUAL | 2 |
| YES | YES | 2 * HIGH_QUAL <= this_qual + pair_qual | 12 |
| YES | YES | 2 * MODERATE_QUAL <= this_qual + pair_qual < 2 * HIGH_QUAL | 10 |
| YES | YES | 2 * LOW_QUAL <= this_qual + pair_qual < 2 * MODERATE_QUAL | 8 |
| YES | YES | this_qual + pair_qual < 2 * LOW_QUAL | 6 |
| YES | NO | HIGH_QUAL <= this_qual - pair_qual | 5 |
| YES | NO | MODERATE_QUAL <= this_qual - pair_qual < HIGH_QUAL | 3 |
| YES | NO | LOW_QUAL <= this_qual - pair_qual < MODERATE_QUAL | 1 |
| YES | NO | this_qual - pair_qual < LOW_QUAL | 0 |
In this table:
this_qualis the quality of this basepair_qualis the quality of the corresponding in the overlapped region of a pair.HIGH_QUALis the quality threshold that can be specified by--high_qualMODERATE_QUALis the quality threshold that can be specified by--moderate_qualLOW_QUALis the quality threshold that can be specified by--low_qual
In the overlapped region, if a base and its pair are mismatched, its quality score will be adjusted to: max(0, this_qual - pair_qual)
If you want to get very clean data, we can only keep the clusters with 2 or more supporting reads (recommended for ultra-deep sequencing with higher dup-rate):
gencore -i in.bam -o out.bam -r hg19.fa -s 2
If you want to keep all the DNA fragments, we can set supporting_reads to 1 (this option can be used to replace picard markduplicate to deduplication):
gencore -i in.bam -o out.bam -r hg19.fa -s 1
(Recommanded) If you want to keep all the DNA fragments, and for each output read you want to discard all the low quality unoverlapped mutations to obtain a relative clean data (recommended for dup-rate < 50%):
gencore -i in.bam -o out.bam -r hg19.fa -s 1 --score_threshold=9
If you want to obtain fewer but ultra clean data, and your data has UMI, you can enable the duplex_only option, and increase the supporting_reads and the ratio_threshold:
gencore -i in.bam -o out.bam -r hg19.fa --duplex_only -s 3 --ratio_threshold=0.9
Please note that only UMI-integrated paired-end data can be used to generate duplex consensuses sequences.
gencore supports calling consensus reads with or without UMI. Although UMI is not required, it is strongly recommended. If your FASTQ data has UMI integrated, you can use fastp to shift the UMI to read query names.
The UMI should in the tail of query names. It can have a prefix like UMI, followed by an underscore. If the UMI has a prefix, it should be specified by --umi_prefix or -u. If the UMI prefix is umi or UMI, it can be automatically detected. The UMI can also have two parts, which are connected by an underscore.
- Read query name =
"NB551106:8:H5Y57BGX2:1:13304:3538:1404:UMI_GAGCATAC", prefix ="UMI", umi ="GAGCATAC" - Read query name =
"NB551106:8:H5Y57BGX2:1:13304:3538:1404:umi_GAGC_ATAC", prefix ="umi", umi ="GAGC_ATAC" - Read query name =
"NB551106:8:H5Y57BGX2:1:13304:3538:1404:GAGCATAC", prefix ="", umi ="GAGCATAC" - Read query name =
"NB551106:8:H5Y57BGX2:1:13304:3538:1404:GAGC_ATAC", prefix ="", umi ="GAGC_ATAC"
options:
-i, --in input sorted bam/sam file. STDIN will be read from if it's not specified (string [=-])
-o, --out output bam/sam file. STDOUT will be written to if it's not specified (string [=-])
-r, --ref reference fasta file name (should be an uncompressed .fa/.fasta file) (string)
-b, --bed bed file to specify the capturing region, none by default (string [=])
-x, --duplex_only only output duplex consensus sequences, which means single stranded consensus sequences will be discarded.
--no_duplex don't merge single stranded consensus sequences to duplex consensus sequences.
-u, --umi_prefix the prefix for UMI, if it has. None by default. Check the README for the defails of UMI formats. (string [=auto])
-s, --supporting_reads only output consensus reads/pairs that merged by >= <supporting_reads> reads/pairs. The valud should be 1~10, and the default value is 1. (int [=1])
-a, --ratio_threshold if the ratio of the major base in a cluster is less than <ratio_threshold>, it will be further compared to the reference. The valud should be 0.5~1.0, and the default value is 0.8 (double [=0.8])
-c, --score_threshold if the score of the major base in a cluster is less than <score_threshold>, it will be further compared to the reference. The valud should be 1~20, and the default value is 6 (int [=6])
-d, --umi_diff_threshold if two reads with identical mapping position have UMI difference <= <umi_diff_threshold>, then they will be merged to generate a consensus read. Default value is 1. (int [=1])
-D, --duplex_diff_threshold if the forward consensus and reverse consensus sequences have <= <duplex_diff_threshold> mismatches, then they will be merged to generate a duplex consensus sequence, otherwise will be discarded. Default value is 2. (int [=2])
--high_qual the threshold for a quality score to be considered as high quality. Default 30 means Q30. (int [=30])
--moderate_qual the threshold for a quality score to be considered as moderate quality. Default 20 means Q20. (int [=20])
--low_qual the threshold for a quality score to be considered as low quality. Default 15 means Q15. (int [=15])
--coverage_sampling the sampling rate for genome scale coverage statistics. Default 10000 means 1/10000. (int [=10000])
-j, --json the json format report file name (string [=gencore.json])
-h, --html the html format report file name (string [=gencore.html])
--debug output some debug information to STDERR.
--quit_after_contig stop when <quit_after_contig> contigs are processed. Only used for fast debugging. Default 0 means no limitation. (int [=0])
-?, --help print this message
The gencore paper has been published in BMC Bioinformatics: https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3280-9. If you used gencore in your research work, please cite it as:
Chen, S., Zhou, Y., Chen, Y. et al. Gencore: an efficient tool to generate consensus reads for error suppressing and duplicate removing of NGS data. BMC Bioinformatics 20, 606 (2019) doi:10.1186/s12859-019-3280-9