{kind=link}
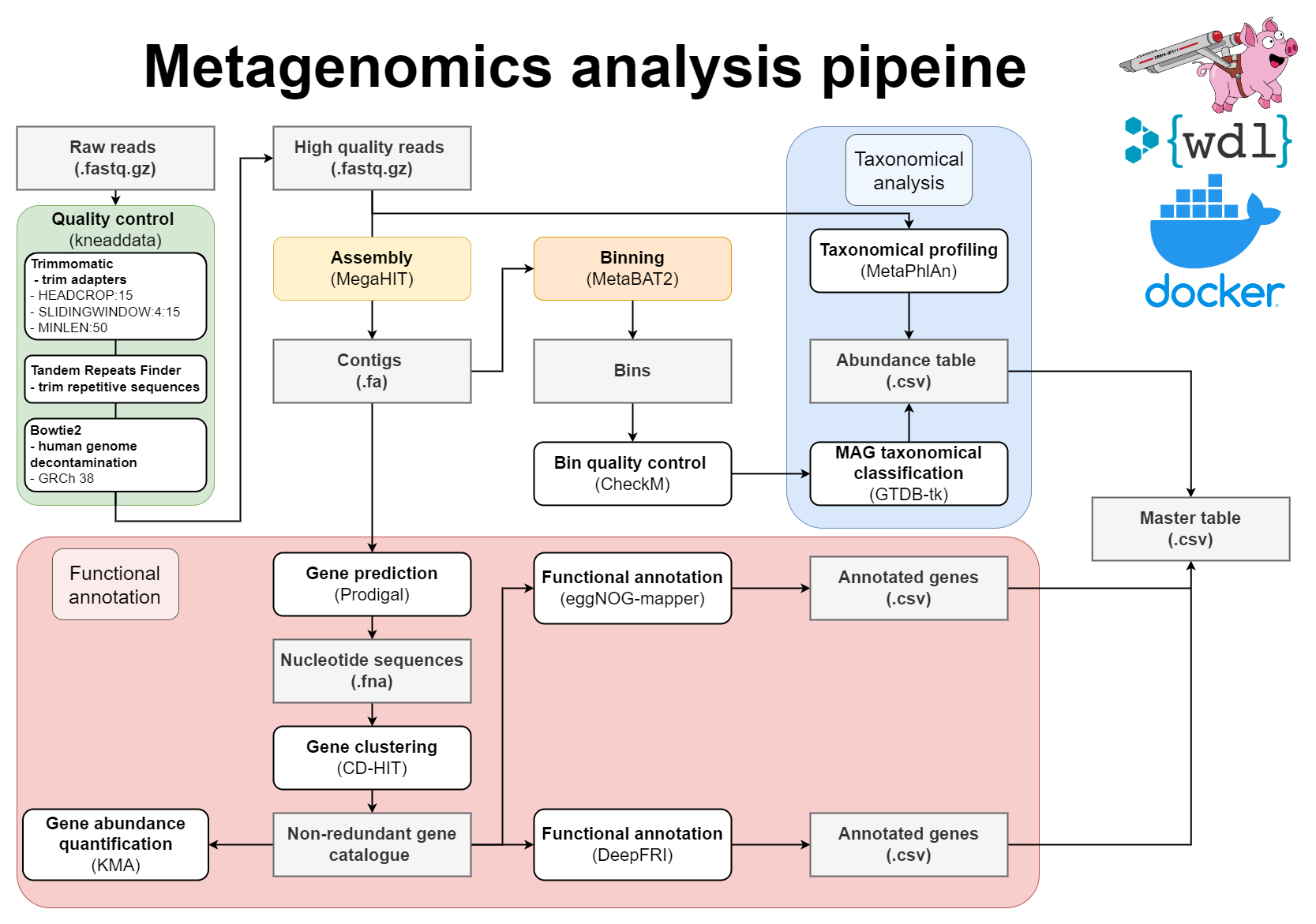
WDL Workflow for metagenome assembly:
 Python script to generate a mapping between non-redundant gene catalog and MAGS
Python script to generate a mapping between non-redundant gene catalog and MAGS
The wrapper scripts in Python (located in src) will prepare files and send them to Cromwell. Cromwell executes instructions written in Workflow definition Language (WDL; located in src/wdl). To avoid dependency conflicts Cromwell runs Docker containers with preinstalled software (dockerfiles located in docker).
Cromwell is an open-source workflow manager for scientific workflows written in WDL. It is designed for handling large-scale genomic data analysis and provides features such as workflow branching, looping, and integration with other systems. It can be run on various platforms including cloud platforms. More information can be found in the documentation: https://cromwell.readthedocs.io/en/stable/
- Pre-processing of reads with Kneaddata
- Metagenomics assembly with Megahit
- Gene prediction
- Mapping of reads against the contigs
- Metagenome binning using MetaBAT2
- Quality assessment of genome bins
- Taxonomic classifications
- Gene clustering with CD-HIT-EST
- Mapping of reads to gene clusters and computing gene counts
- Functional annotation of proteins using both eggNOG-mapper and DeepFRI
- Mapping between MAGs and functionally annotated gene catalogue
Dockercondafor building the environment- Python
git clone github.com/bioinf-mcb/metagenome_assembly
conda env create -f pipeline.yml
Use the setup_cromwell.py script to download and install it.
- python src/setup_cromwell.py --save_path SAVE_PATH
This step will perform quality control of your reads with BBTools according to Reads QC Workflow v. 1.0.1.
- Requirements
input_folder- path to directory with paired shotgun sequencing files. (fastq.gz,fastq,fq.gz,fq)db_path- path to a directory with an RQCFilter Data. In case the folder doesn't contain a database, it would be downloaded to the path.output_folder- path to a directory where the results will be saved.
- Optional arguments
thread_num- number of threads to use. (default: 1)concurrent_jobs- number of concurrent jobs to run. (default: 1)memory- RAM memory to be used in GB. (default: 60)
- Output
- quality controlled interleaved .fastq.gz file in
OUTPUT_FOLDER/SAMPLE/SAMPLE.anqdpht.fastq.gz- used for assembly - quality controlled paired fastq.gz giles in
OUTPUT_FOLDER/SAMPLE/SAMPLE_paired_1.fastq.gz&OUTPUT_FOLDER/SAMPLE/SAMPLE_paired_1.fastq.gz- used for taxonomical profiling withMetaPhlanormOTUs - QC stats
- quality controlled interleaved .fastq.gz file in
- Resources
- Disk space: 106 GB for the RQCFilterData database
- Memory: ~60 GB RAM
# Qualirty control raw reads and assemble contigs
python src/qc.py -i INPUT_FOLDER -o OUTPUT_FOLDER -t 16 -c 1 -db_path databases/refdataDEBUG:root:Creating output directory: tests/qc
DEBUG:root:Creating output directory: tests/qc/system
INFO:root:Treating /storage/TomaszLab/vbez/metagenomic_gmhi/metagenomome_assembly/databases as directory with RCQFilterData Database.
INFO:root:I inferred that _1 and _2 distinguish paired end reads.
INFO:root:Found samples: 2This step will perform genome assembly with MEGAHIT.
- Requirements
input_folder- path to directory with interleaved sequencing file. (`)output_folder- path to a directory where the results will be saved.
- Optional arguments
thread_num- number of threads to use. (default: 1)concurrent_jobs- number of concurrent jobs to run. (default: 1)
- Output
- contigs filtered by minimum length
- scaffold filtered by minimum length
- QC stats
- Resources
- Memory: ~60 GB RAM
# Qualirty control raw reads and assemble contigs
python src/assemble.py -i INPUT_FOLDER -o OUTPUT_FOLDER -min_len 500 -t 24 -c 1DEBUG:root:Creating output directory: tests/assemble
DEBUG:root:Creating output directory: tests/assemble/system
[11:32:03] Workflow assemble has started. Please, be patient.
[11:34:54] Workflow finished successfully.This step will bin contigs using MetaBAT2, check bins for quality and contamination using CheckM and assign taxonomical classification for MAGs using GTDB.
- Requirements
input_folder_reads- a path to a directory with QCed reads (located inOUTPUT_FOLDER/ofqc and assemblystep).input_folder_contigs- a path to a directory with assembled contigs (located inOUTPUT_FOLDER/assembleofqc and assemblystep).gtdbtk_data- a path to a directory with a GTDB-Tk database release. In case the folder doesn't contain the data, it will be downloaded automatically.output_folder- a path to a directory where the results will be saved.
- Optional arguments
thread_num- number of threads to use. (default: 1)concurrent_jobs- number of concurrent jobs to run. (default: 1)suffix- suffix, that helps to identify contigs and preserve consistent filenames. (default:.min500.contigs.fa)suffix1- suffix, that helps to identify forward reads. (default:_paired_1.fastq.gz)suffix2- suffix, that helps to identify reverse reads. (default:_paired_2.fastq.gz)
- Output
SAMPLE_NAME.gff- feature table in Genbank table.SAMPLE_NAME.fna- nucleotide sequences for genes in FASTA.SAMPLE_NAME.faa- protein translations for genes in FASTA.
# Bin, check and taxonomically classify MAGs
python src/t1_predict_mags.py -ir INPUT_FOLDER_READS -s1 _paired_1.fastq.gz -s2 -s1 _paired_2.fastq.gz \
-ic INPUT_FOLDER_CONTIGS -s .min500.contigs.fa \
-gtdb ../databases/gtdbtk-data/ -o OUTPUT_FOLDER \
-t 24 -c 2DEBUG:root:Creating output directory: out_mags
DEBUG:root:Creating output directory: out_mags/system
[17:23:29] Workflow started succesfully. Please, be patient.
[17:40:13] Workflow finished successfully.
This step will perform gene recognition using Prodigal.
- Requirements
input_folder- a path to a directory with assembled contigs (located inOUTPUT_FOLDER/assembleof theqc and assemblystep).output_folder- a path to a directory where the results will be saved.
- Optional arguments
concurrent_jobs- number of concurrent jobs to run. (default: 1)suffix- suffix, that helps to identify contigs and preserve consistent filenames. (default:.min500.contigs.fa)
- Output
SAMPLE_NAME.gff- feature table in Genbank table.SAMPLE_NAME.fna- nucleotide sequences for genes in FASTA.SAMPLE_NAME.faa- protein translations for genes in FASTA.
# Predict genes
python src/f1_predict_genes.py -i INPUT_FOLDER -s .min500.contigs.fa -o OUTPUT_FOLDER \
-c 3DEBUG:root:Creating output directory: OUTPUT_FOLDER
DEBUG:root:Creating output directory: OUTPUT FOLDER/system
[15:19:43] Workflow started succesfully. Please, be patient.
[15:21:29] Workflow finished successfully.This step will cluster genes using CD-HIT and sequence similarity threshold.
- Requirements
input_folder- a path to a directory with predicted nucleotide sequences of genes (OUTPUT_FOLDER/*.fnaof the previous step).output_folder- a path to a directory where the results will be saved.
- Optional arguments
thread_num- number of threads. (default: 1)suffix- suffix, that helps to identify contigs and preserve consistent filenames. (default:.fna)
- Output
gene_catalogue_split- gene cataloge split in chunks of 10,000 sequences for further analysis.combined_genepredictions.sorted.fna- combined predictions of complete genes sorted by length.nr.fa- full gene catalogue.nr.fa.clstr- clustered genes.kma_db.tar.gz- KMA database - required for quantification of gene copies in bacterial genomes (next step).
# Cluster genes
python src/f2_generate_gene_catalog.py -i INPUT_FOLDER -s .fna -o OUTPUT_FOLDER \
-t 16DEBUG:root:Creating output directory: OUTPUT_FOLDER
DEBUG:root:Creating output directory: OUTPUT FOLDER/system
[15:23:26] Workflow started succesfully. Please, be patient.
[15:24:56] Workflow finished successfully.This step will quantify the number of gene clusters in sequenced reads aligning it to the reference using KMA.
- Requirements
input_folder- a path to a directory with quality-controlled reads (from theqc_and_assemblystep).database- a path to a KMA database. (fromF2 - Gene clusteringstep)output_folder- a path to a directory where the results will be saved.
- Optional arguments
suffix1- suffix, that helps to identify forward reads. (default:_paired_1.fastq.gz)suffix2- suffix, that helps to identify reverse reads. (default:_paired_2.fastq.gz)thread_num- number of threads. (default: 1)
- Output
SAMPLE_NAME.kma.res- KMA full output.SAMPLE_NAME.geneCPM.txt- table with extracted and normalized gene counts (count per million).
# Quantify gene clusters
python src/f3_generate_gene_catalog.py -i INPUT_FOLDER -s1 _paired_1.fastq.gz -s2 _paired_2.fastq.gz \
-db F2_OUTPUT_FOLDER/kma_db.tar.gz \
-o OUTPUT_FOLDER \
-t 16DEBUG:root:Creating output directory: OUTPUT_FOLDER
DEBUG:root:Creating output directory: OUTPUT FOLDER/system
[15:26:48] Workflow started succesfully. Please, be patient.
[15:29:08] Workflow finished successfully.This step will provide functional annotation of gene clusters from both eggNOG-mapper and DeepFRI.
- Requirements
input_folder- a path to a directory with gene catalog split into chunks of 10,000 reads (fromF2 - gene clusteringstep).eggnog_database- a path to aneggNOG-mapperdatabase. If it is not located in the folder, the necessary files will be downloaded automatically.output_folder- a path to a directory where the results will be saved.
- Optional arguments
suffix- suffix, that helps to gene catalog chunks. (default:.fa)thread_num_- number of threads. (default: 1)concurrent_jobs- number of jobs to run in parallel. A singleDeepFRIjob requires 55GB of RAM, too many jobs may result in an out-of-memory error.
- Output
deepfri_annotations.csv-DeepFRIfunctional annotation for a gene catalog.nr-eggnog.emapper.annotations-eggNOG-mapperfunctional annotation for a gene catalog.nr-eggnog.emapper.seed_orthologs- a file with the results from parsing the hits. Each row links a query with a seed ortholog.
# Annotate gene catalog
python src/f4_annotate_gene_catalog.py
-i F2_OUTPUT_FOLDER/gene_catalog_split/ -s .fa \
-db eggNOG-DATABASE \
-o OUTPUT_FOLDER \
-t 16 -c 2INFO:root:Treating /storage/TomaszLab/vbez/metagenomic_gmhi/metagenomome_assembly/databases/eggnog-data as directory with eggNOG.
INFO:root:Treating /storage/TomaszLab/vbez/metagenomic_gmhi/metagenomome_assembly/databases/eggnog-data as directory with Diamond.
DEBUG:root:Creating output directory: OUTPUT_FOLDER
DEBUG:root:Creating output directory: OUTPUT_FOLDER/system
[15:56:27] Workflow started succesfully. Please, be patient.
[16:47:31] Workflow finished successfully.
This step will collect all the output into one table.
-
Requirements
contig_folder- a path to a directory with contigs from theqc_and_assemblystep.bins_folder- a path to a directory with bins from theT1 - MAGs binningstep.gtdbtk_folder- a path to a directory with GTDB-Tk results from theT1 - MAGs binningstep.checkm_folder- a path to a directory with CheckM results from theT1 - MAGs binningstep.gene_catalog- a path to a gene catalog file from theF2 - gene clusteringstep.gene_cluster_file- a path to a file with gene clusters.eggnog_annotations- a path to a file witheggNOG-mapperannotations.deepfri_annotations- a path to a file withDeepFRIannotations.output_folder- a path to a directory where the results will be saved.
-
Outputs
_individual_mapped_genes.tsv- genes clusters mapped to MAGs._MAGS.tsv- MAGs summary fromGTDB-tkandCheckM._mapped_genes_cluster.tsv-eggNOG-mapperannotations for gene clusters.merged_eggnog_output.tsv-eggNOG-mapperannotations for gene clusters.merged_deepfri_output.tsv-DeepFRIannotations for gene clusters.
# Generate final output
python ../src/generate_table.py \
-c qc_assemble_out/assemble \
-b t1_output \
-g t1_output \
-cm t1_output \
-gcf f2_output/nr.fa.clstr \
-gc f2_output/nr.fa \
-ea f4_output \
-dfa f4_output \
-o final_outTo get help with the pipeline, please open an issue on the Github tracker.
Repo maintainer: Valentyn Bezshapkin
General inquiries: Tomasz Kosciolek