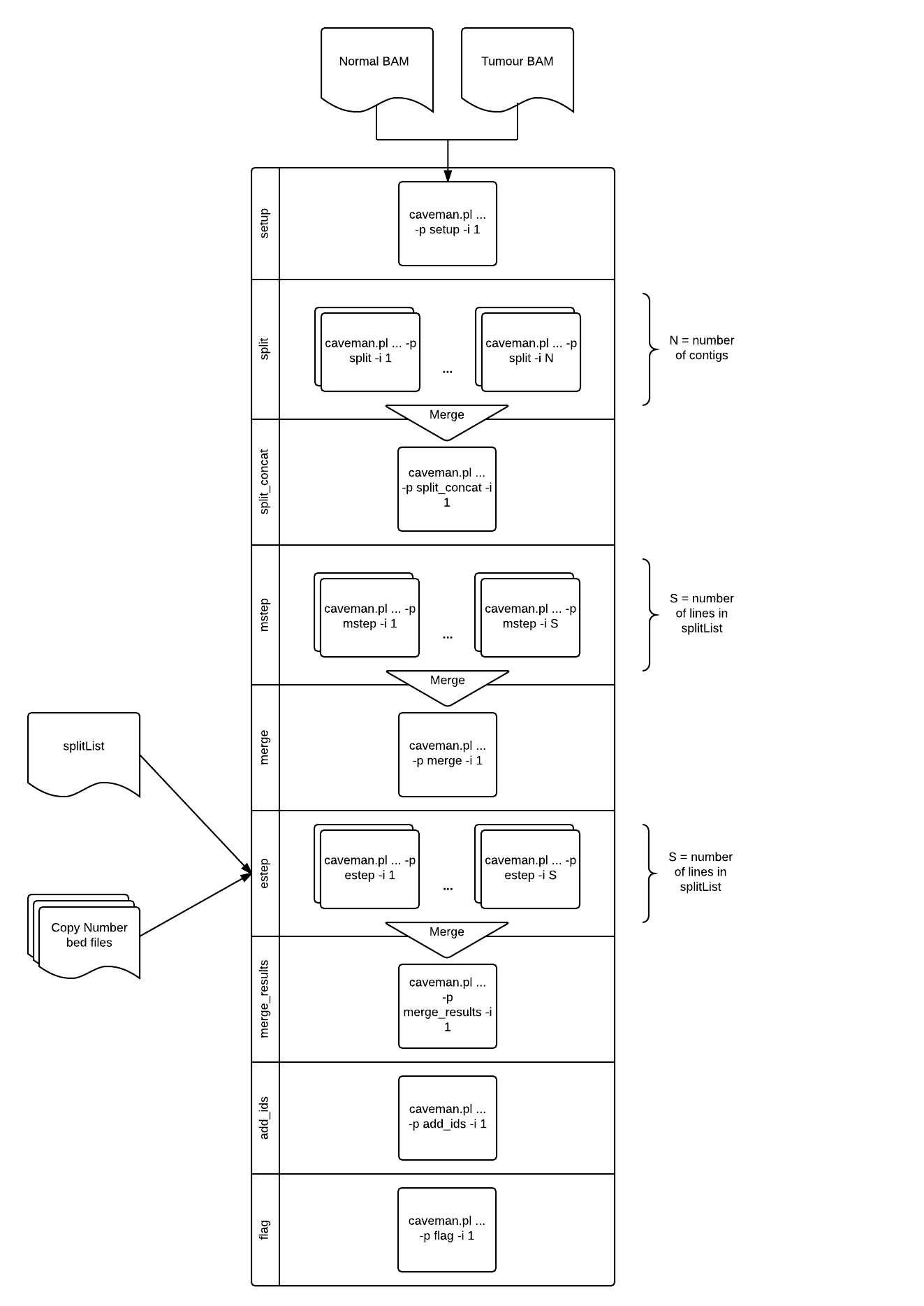
Execution
Option 2 provides more control over individual steps in the process. For CaVEMan option 2 is preferred as option 1 results in much longer runtimes.
Single host execution does require a reasonable level of resources. Minimum specification (Based on WGS at 30x coverage):
- CPU 32
- RAM (Gb) 4 / CPU (Some data may require far higher resources)
NB The code will recover from failure and restart at the last successful section.
For users with access to a compute farm it is possible to break down the execution into the component parts (see Overview).
There are 9 steps:
- setup
- Generate configuration files for the remaining steps.
- Command suffixed with
-process setup -index 1
- split
- Split the genome into chunks by readsize and hard stop forced by contig ends
- Command suffixed with
-process setup -index X -
Xis between1and the number of contigs inreference.fa.fai
- split_concat
- Concatenate the split file sections into a single split section reference file
- Command suffixed with
-process split_concat -index 1 - Generates
splitList
- mstep
- One job per line in
splitList, builds a profile of the section of the genome using various covariates - Command suffixed with
-process mstep -index N -
Nis between1and the number of lines insplitList
- merge
- Merge the mstep profiles into a single profile for the whole genome
- Command suffixed with
-process merge -index 1 - Produces
probs_arrandcovs_arr
- estep
- One job per line in
splitList, uses the profile built in the mstep, combined with sequence data and copy number to assign a probability to each possible genotype at each position. - Command suffixed with
-process estep -index N -
Nis between1and the number of lines insplitList - Positions where germline mutation probabilities total more than the
-snp-cutoffare output to<split_section>.snps.vcf - Positions where somatic mutation probabilities total more than the
-mut-cutoffare output to<split_section>.muts.vcf
- merge_results
- Merges all output results files into per-genome files names
T_vs_N.*.vcf. - Command suffixed with
-process merge_results -index 1
- add_ids
- Appends UUIDs to each entry in both the snps and the muts output VCF files.
- Command suffixed with
-process add_ids -index 1
- flag
- Applies filters to the
T_vs_N.muts.vcf.gzfile. ProducesT_vs_N.muts.flagged.vcf.gz - Command suffixed with
-process flag -index 1
Flagging is described in further detail on the cgpCaVEManPostProcessing wiki