User manual
$ pip install git+https://github.com/huang-sh/raatk.git@master -U
or
$ pip install raatk
- view view reduced amino acid alphabet
- reduce reduce amino acid sequence
- extract extract sequence feature
- hpo hpyper-parameter optimization
- eval evaluation
- plot result visualization
- roc ROC evaluation
- ifs feature selection
- train train model
- predict prediction
- split split data
- transfer transfer format
view is used to view build-in reduced amino acid alphabets. And for online browsing, please visit http://bioinfor.imu.edu.cn/raacbook/public/
Arguments:
- -t: reduced amino acid alphabets id
- -s: reduced amino acid cluster size
- --visual: for visualization
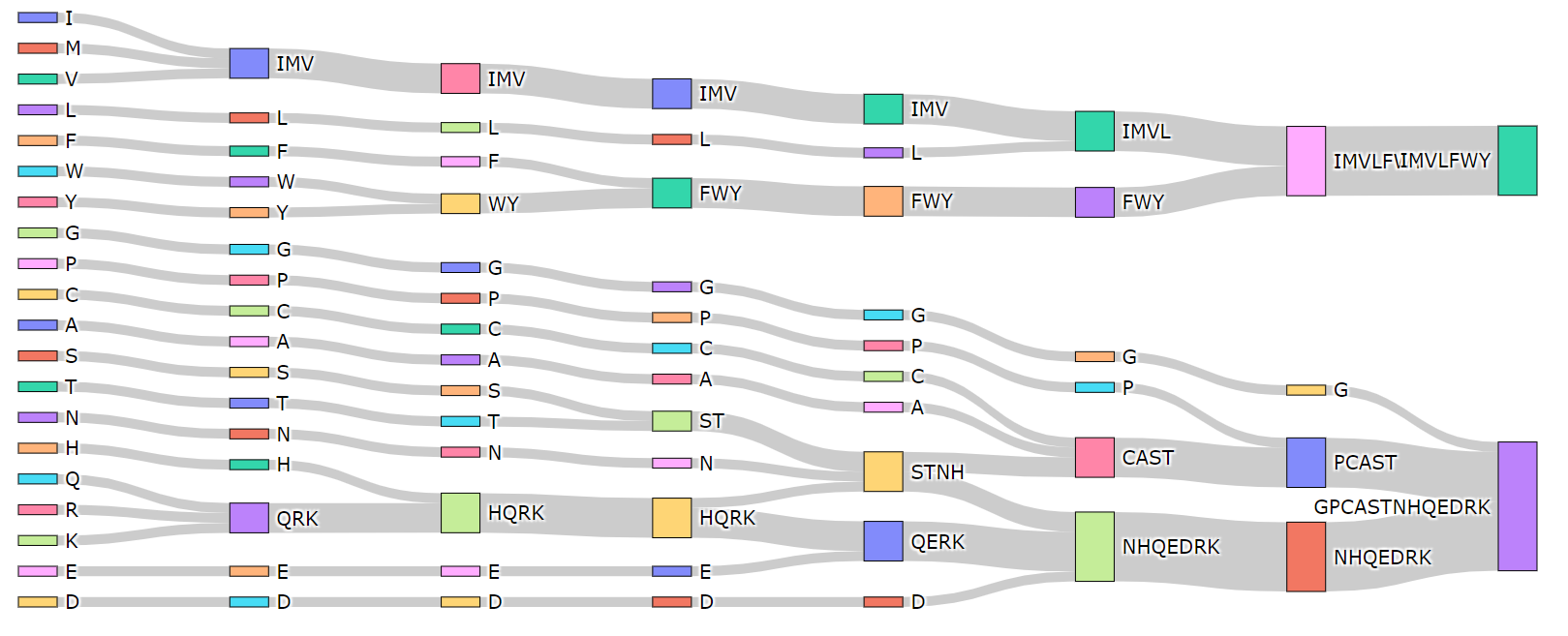
$raatk view -t 9 -s 2 4 6 10 12 14 16 --visualOutput:
type9 2 IMVLFWY-GPCASTNHQEDRK BLOSUM50 matrix
type9 4 IMVLFWY-G-PCAST-NHQEDRK BLOSUM50 matrix
type9 6 IMVL-FWY-G-P-CAST-NHQEDRK BLOSUM50 matrix
type9 10 IMV-L-FWY-G-P-C-A-STNH-QERK-D BLOSUM50 matrix
type9 12 IMV-L-FWY-G-P-C-A-ST-N-HQRK-E-D BLOSUM50 matrix
type9 14 IMV-L-F-WY-G-P-C-A-S-T-N-HQRK-E-D BLOSUM50 matrix
type9 16 IMV-L-F-W-Y-G-P-C-A-S-T-N-H-QRK-E-D BLOSUM50 matrix
reduce sequence according to built-in or user-defined reduction alphabets.
Arguments:
- file: positional argument, the input amino acid fasta file
- -t: reduced amino acid alphabets id
- -s: reduced amino acid cluster size
- -c: user-defined reduced cluster
- -naa: if set, the output will include original sequence
- -o: output path
If you use built-in reduction alphabets, you should set -t and -s. And the output is stored in directories.
$raatk reduce positive.txt negative.txt -t 1-8 -s 2-19 -o pos negIf you use customized reduction alphabet, you should set -c argument. And the output result is in file.
$raatk reduce positive.txt -c IMV-L-FWY-G-P-C-A-STNH-QERK-D -o reduce_positive.txtextract the features of amino acid sequences according to relevant parameters.
Arguments:
- file: positional argument, input fasta sequence file or directory
- -d: when file argument input is directory, you should set this
- -k: k-mer or k-tuple
- -g: gap value
- -l: lambda-correlation value
- -raa: when file argument input is reduced sequence file, you should set -raa with representative amino acid. For example, if the input file is reduce by 'IMV-L-FWY-G-P-C-A-STNH-QERK-D', you should set -raa 'ILFGPCASQD'
- -idx: feature indexes, used to extract features with specific indexes
- -m: when multiple files are extracted, this can merge them into one
- -o: output path
- -p: Number of processor
- --label-f: output feature file without label
- --count: feature count
extract sequence features of directories, and the output is also stored in directory.
$raatk extract pos neg -k 3 -d -o k3 -mextract sequence features of files, and the output is also stored in file.
$raatk extract pos/type9/4-IGPN.txt neg/type9/4-IGPN.txt -k 1 -o t9s4-k1.csv -m -raa IGPNOutput:
label,I,G,P,N
0.000000,0.125000,0.062500,0.562500,0.250000
0.000000,0.291667,0.166667,0.416667,0.125000
0.000000,0.277778,0.083333,0.416667,0.222222
......
1.000000,0.177778,0.133333,0.377778,0.311111
1.000000,0.166667,0.000000,0.583333,0.250000
1.000000,0.387097,0.161290,0.322581,0.129032
feature file without label and count the features
$raatk extract pos/type9/4-IGPN.txt -k 1 -o t9s4-k1p.csv -raa IGPN --count --label-fOutput:
I,G,P,N
2.000000,1.000000,9.000000,4.000000
7.000000,4.000000,10.000000,3.000000
10.000000,3.000000,15.000000,8.000000
......
hpo is a command for hpyper-parameter optimization using grid search method.
Arguments:
- file: positional argument, feature file for hpyper-parameter optimization
- -clf: classifier, default is svm
- -jobs: number of parallel jobs to run, default=1
- -c: regularization parameter for SVM, format: [start stop [number]]. For example: -c -2 2 5, and it will be transform to 2^-2,2^-1,2^0,2^1,2^2
- -g: gamma,kernel coefficient for SVM, format: [start stop [number]]. For example: -c -2 2 5, and it will be transform to 2^-2,2^-1,2^0,2^1,2^2
- -k: SVM kernel, options: {rbf,linear}
$raatk hpo k3/type2/10-ARNCQHIFPW.csv -c -5 5 11 -g -5 3 9 -jobs 3 -k rbf linear
C: 2.0, gamma: 1.0, kernel: rbf
evaluate the performance of different alphabet clusters based on machine learning.
Arguments:
- file: positional argument, feature file or directory
- -d: if set, the file argument value is directory path
- -clf: classifier model. svm, rf or knn can be set and default is svm
- -cv: stratified K-Folds cross-validation. when -1 is set, this is leave-one-out cross-validation
- -o: output path
- -p: number of processor classifier model parameters could be viewed by:
$raatk eval -hevaluate the different reduced feature files in directory. And the output is a json file.
$raatk eval k3 -d -o k3-eval -clf svm -c 2 -g 0.5 -p 3evaluate a single file.
$raatk eval k3/type2/10-ARNCQHIFPW.csv -cv -1 -c 2 -g 0.5 -o k3-t2s10.txtoutput:
0
0 38 7
1 7 36
tp fn fp tn recall precision f1-score
0 38 7 7 36 0.84 0.84 0.84
1 36 7 7 38 0.84 0.84 0.84
acc 0.84
mcc 0.68
-------------------------------------------------------
json file visualization
Arguments:
- file: json file path
- -fmt: output image format, options are: pdf,png,eps,ps,raw,rgba,svg
- -o: output directory
$raatk plot k3-eval.json -o k3poutput:
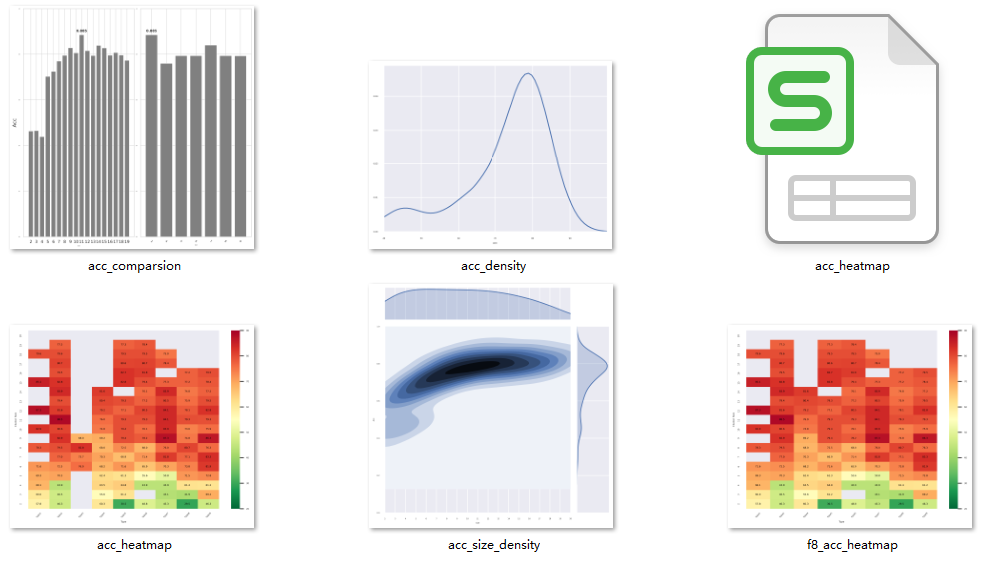
- acc_heatmap.png: it is a heatmap made by using all the ACC values obtained by evaluating the reduced amino acid alphabet. The abscissa is the type id of different reduced alphabets, indicating different amino acid reduction method; the ordinate is the size of the reduced amino acid alphabet cluster, which is reduced from the natural 20 types of amino acids to a smaller number of classes
- acc_heatmap.csv: it is csv format version of acc_heatmap.png
- f8_acc_heatmap: Similar to acc_heatmap.png, but filter types that are less than 8 sizes
- acc_comparsion: On the left of the figure, it is the bar chart of ACC evaluation with different Cluster sizes within the same type; On the right of the figure, it is a bar chart for evaluating ACC with different types and the same size (f8_acc_heatmap data is used)
- acc_density: use ACC value of f8_acc_heatmap to fit and plot a univariate gaussian kernel density estimate
- acc_size_density: use data of f8_acc_heatmap to fit and plot a bivariate gaussian kernel density estimate.
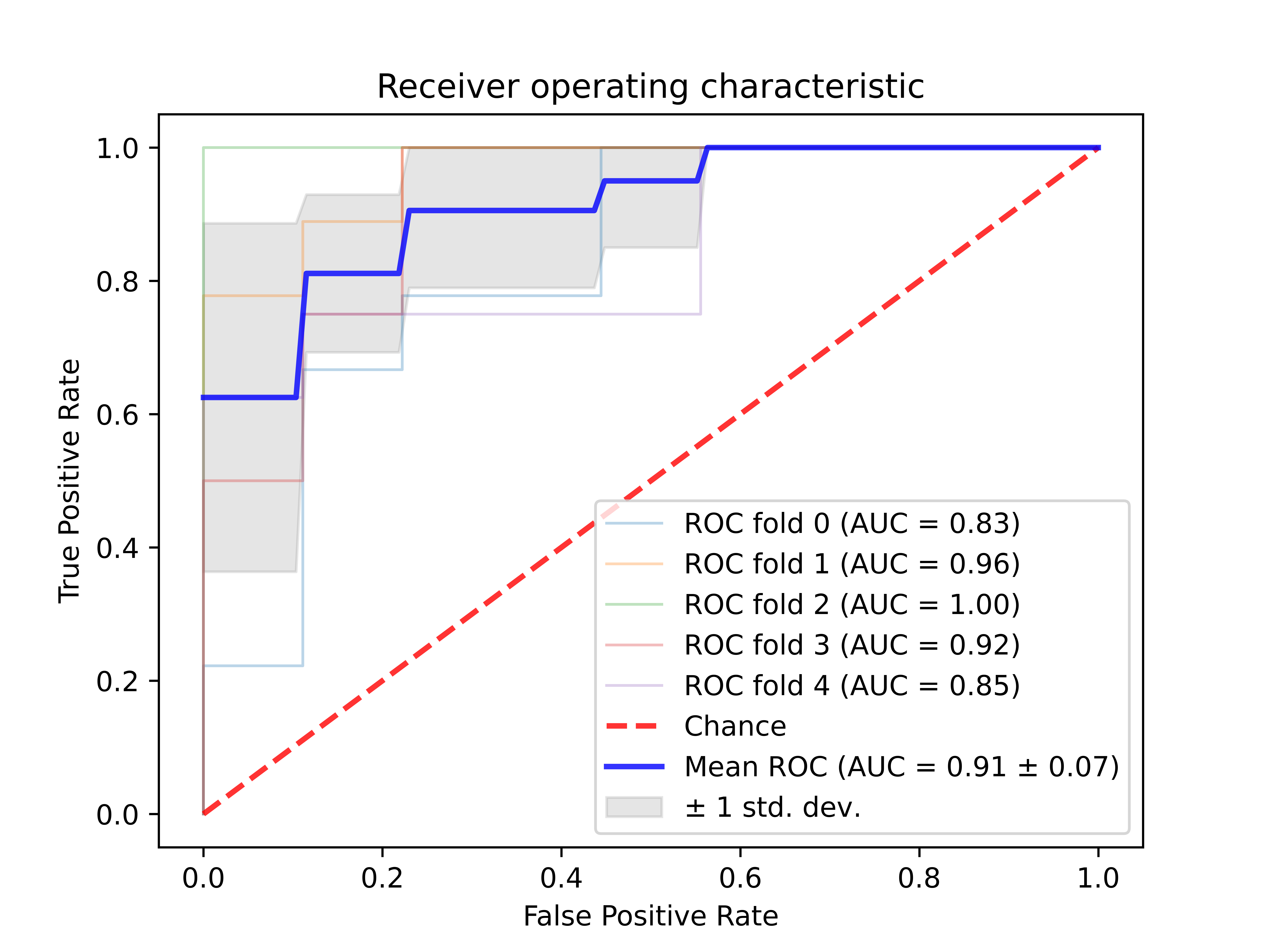
trained classifier model ROC evaluation or classifier algorithm ROC evaluation by cross validation
Arguments:
- file: feature file for ROC evaluation
- m: trained model
- -clf: classifier model. svm, rf or knn can be set
- -cv: stratified K-Folds cross-validation. when -1 is set, this is leave-one-out cross-validation
- -fmt: output image format, options are: {eps,pdf,png,ps,raw,rgba,svg,txt}. when txt is set, a csv output is generated and user can use it for new plot.
- -o: output
$raatk roc k3/type2/10-ARNCQHIFPW.csv -clf svm -cv 5 -c 2 -g 0.5 -o rocoutput:
Incremental feature selection. Use ANOVA to compute f-score for each feature, then rank features according to their f-score from large to small. First, compute Acc with first S features, then compute first 2S features and until all feature is computed(S >= 1) The features corresponding to the highest ACC are the optimal features.
Arguments:
- file: feature file for feature selection
- -s:step, the incremental features number
- -clf: classifier model. svm, rf or knn can be set
- -cv: stratified K-Folds cross-validation. when -1 is set, this is leave-one-out cross-validation
- -o: output
- -p: number of processor
$raatk ifs k3/type2/10-ARNCQHIFPW.csv -s 2 -clf svm -cv 5 -c 2 -g 0.5 -o ifsoutput:
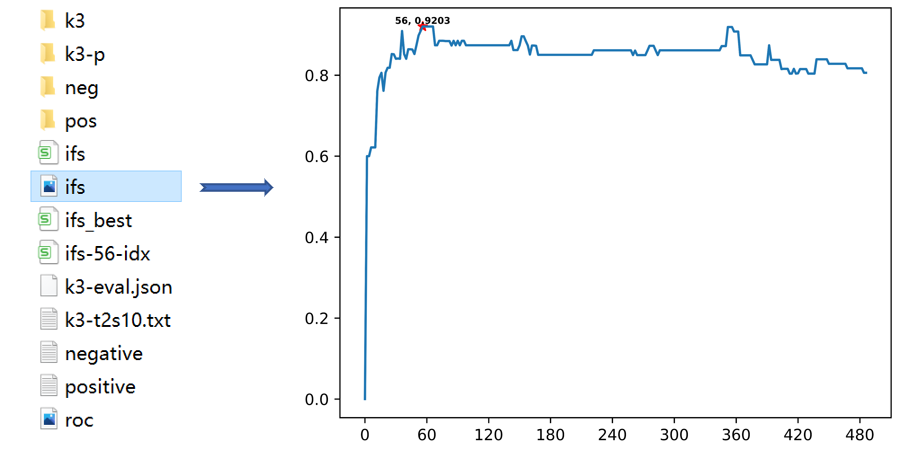
- ifs.png: feature selection figure
- ifs.csv: csv format file of ifs.png. it can be used to draw new figure
- ifs_best.csv: the features corresponding to the highest ACC value, it can be used to train the model
- ifs_56-idx.csv: the feature indexes corresponding to the highest ACC value。 It can used to extract to specific feature with help of extract -idx parameter
train a classifier model
Arguments:
- file: feature file
- -clf: classifier model. svm, rf or knn can be set
- -o: output
$raatk train ifs_best.csv -clf svm -c 2 -g 0.5 -o svm.model -probpredict new data using trained model. The new data must be feature file without label and feature extract parameter must be same as training feature.
Arguments:
- file: feature file
- -m: trained model
- -o: output
$raatk predict new_data.csv -m svm.model -o 'test-result.csv'split feature data into train and test subsets
Arguments:
- file: feature file
- -ts: test set size,float,0<ts<1
- -o: output
$raatk split ifs_best.csv -ts 0.3 -o test_split.csvtransfer csv to arff for Weka.
Arguments:
- file: feature file
- -fmt: transfer format, support arff
$raatk transfer ifs_best.csv -fmt arff