![]()
![]()
![]()

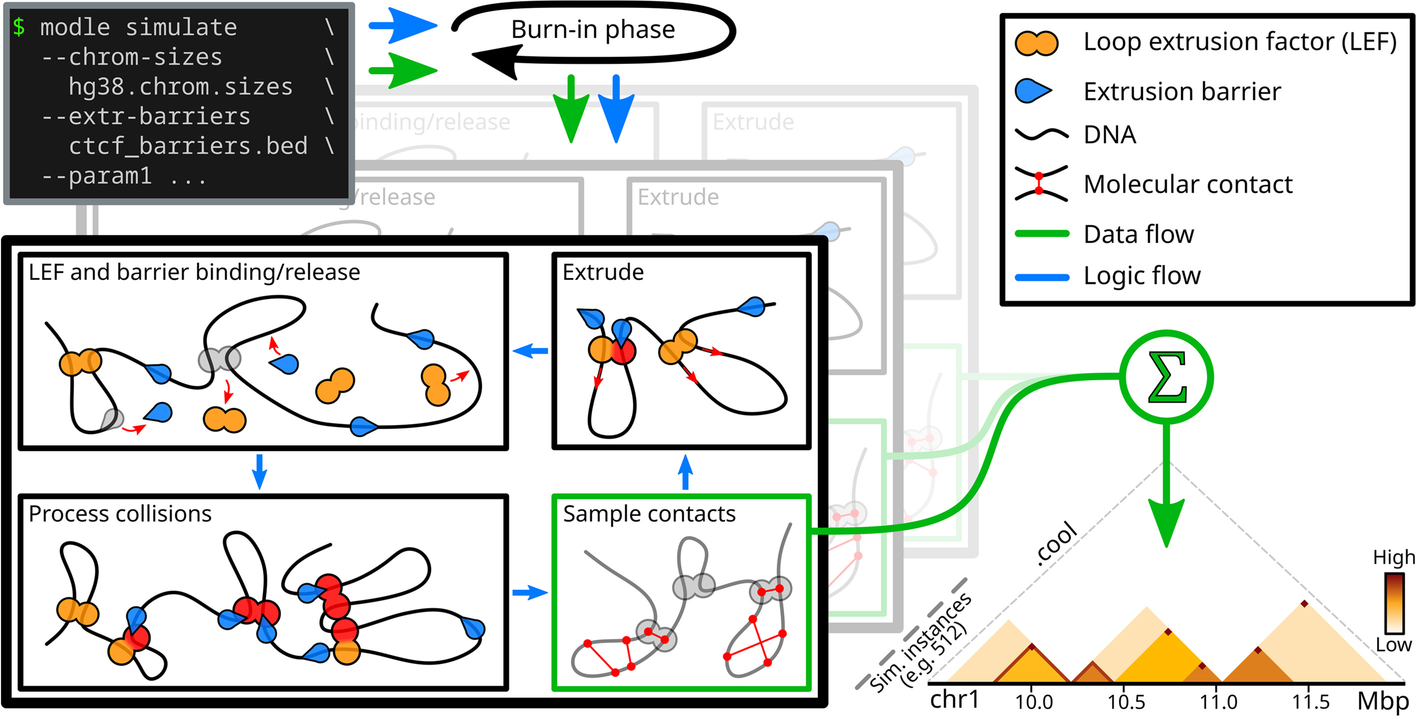
MoDLE is a computational tool for fast, stochastic modeling of molecular contacts from DNA loop extrusion capable of simulating realistic contact patterns genome wide in a few minutes.
MoDLE is developed on Linux and tested on Linux and MacOS. Running MoDLE on Windows is only possible using Docker containers (see below).
MoDLE can be installed or compiled with one of the following methods.
First, make sure you follow the instructions on how to install Docker or Singularity/Apptainer on your OS.
Installing Docker
The following instructions assume you have root/admin permissions.
On some Linux distributions just installing Docker is not enough. You also need to start (and optionally enable) the appropriate service(s). This is usually done with one of the following:
sudo systemctl start docker
sudo systemctl start docker.serviceRefer to Docker or your distribution documentation for more details.
Installing Singularity/Apptainer
The following instructions assume you have root/admin permissions.
Apptainer can be easily installed using your system package manager.
Here you can find instructions for common Linux distributions such as Ubuntu.
Even if your distribution is not listed in the above documentation, your system package manager likely includes a package for Singularity or Apptainer. If this is not the case, then you must install Apptainer from source (instructions available here).
MoDLE Docker images are available on ghcr.io and dockerhub.
Downloading and running the latest stable release can be done as follows:
# Using Docker, may require sudo
user@dev:/tmp$ docker run ghcr.io/paulsengroup/modle:1.1.0 --help
# Using Singularity/Apptainer
user@dev:/tmp$ singularity run ghcr.io/paulsengroup/modle:1.1.0 --help
High-performance stochastic modeling of DNA loop extrusion interactions.
Usage: /usr/local/bin/modle [OPTIONS] SUBCOMMAND
Options:
-h,--help Print this help message and exit
-V,--version Display program version information and exit
--config Path to MoDLE's config file (optional).
Subcommands:
simulate, sim Simulate loop extrusion and write resulting molecular contacts in a .cool file.The above will print MoDLE's help message, and is equivalent to running modle --help on the command line (assuming MoDLE is available on your machine).
Troubleshooting
Errors regarding missing input file(s)
Assuming you have placed the required input files inside a folder named mydata
user@dev:/tmp$ ls -lah mydata
total 248K
drwx------ 2 user group 80 Jan 5 12:00 .
drwxrwxrwt 20 user group 540 Jan 5 12.00 ..
-rw------- 1 user group 365 Jan 5 12:00 grch38.chrom.sizes
-rw------- 1 user group 241K Jan 5 12:00 grch38_h1_extrusion_barriers.bed.xzRunning MoDLE using Docker as shown below fails due to missing input file(s)
docker run ghcr.io/paulsengroup/modle:1.1.0 simulate \
--chrom-sizes mydata/grch38.chrom.sizes \
--extrusion-barrier-file mydata/grch38_h1_extrusion_barriers.bed.xz \
--output-prefix mydata/output
# Error message
# --chrom-sizes: File does not exist: mydata/grch38.chrom.sizes
# Run with --help for more information.This issue is caused by Docker not mounting/binding the host filesystem (e.g. the filesystem where mydata/ resides) inside the container.
To make mydata/ accessible from inside the container, use one of the following methods
# Method 1: mydata/ folder on the host is mapped to /mydata (notice the /) inside the container
docker run -v "$(pwd -P)/mydata/:/data/" \
ghcr.io/paulsengroup/modle:1.1.0 simulate \
--chrom-sizes /data/grch38.chrom.sizes \
--extrusion-barrier-file /data/grch38_h1_extrusion_barriers.bed.xz \
--output-prefix /data/output
# Mehtod 2: use two different folders for input/output. Input folder is mounted in read-only mode
docker run -v "$(pwd -P)/mydata/:/input_data/:ro" \
-v "$(pwd -P)/output/:/output_data/" \
ghcr.io/paulsengroup/modle:1.1.0 simulate \
--chrom-sizes /input_data/grch38.chrom.sizes \
--extrusion-barrier-file /input_data/grch38_h1_extrusion_barriers.bed.xz \
--output-prefix /output_data/output
# Methods for Singularity/Apptainer (note that manually mounting paths is usually not required when using Singularity/Apptainer)
singularity run -B "$(pwd -P)/mydata/:/data/" \
docker://ghcr.io/paulsengroup/modle:1.1.0 simulate \
--chrom-sizes /data/grch38.chrom.sizes \
--extrusion-barrier-file /data/grch38_h1_extrusion_barriers.bed.xz \
--output-prefix /data/output
singularity run -B "$(pwd -P)/mydata/:/input_data/:ro" \
-B "$(pwd -P)/output/:/output_data/" \
docker://ghcr.io/paulsengroup/modle:1.1.0 simulate \
--chrom-sizes /input_data/grch38.chrom.sizes \
--extrusion-barrier-file /input_data/grch38_h1_extrusion_barriers.bed.xz \
--output-prefix /output_data/outputOutput produced by Method 1:
user@dev:/tmp$ ls -lah mydata
total 57M
drwx------ 2 user group 140 Jan 5 12:20 .
drwxrwxrwt 20 user group 560 Jan 5 12:20 ..
-rw------- 1 user group 365 Jan 5 12:00 grch38.chrom.sizes
-rw------- 1 user group 241K Jan 5 12:00 grch38_h1_extrusion_barriers.bed.xz
-rw-r--r-- 1 root root 3.7K Jan 5 12:00 output_config.toml
-rw-r--r-- 1 root root 2.4M Jan 5 12:05 output_lef_1d_occupancy.bw
-rw-r--r-- 1 root root 57M Jan 5 12:05 output.cool
-rw-r--r-- 1 root root 14K Jan 5 12:05 output.logOutput produced by Method 2:
user@dev:/tmp$ ls -lah mydata/
total 57M
drwx------ 2 user group 140 Jan 5 12:20 .
drwxrwxrwt 20 user group 560 Jan 5 12:20 ..
-rw------- 1 user group 365 Jan 5 12:00 grch38.chrom.sizes
-rw------- 1 user group 241K Jan 5 12:00 grch38_h1_extrusion_barriers.bed.xz
user@dev:/tmp$ ls -lah output/
drwx------ 2 user group 140 Jan 5 12:20 .
drwxrwxrwt 20 user group 560 Jan 5 12:20 ..
-rw-r--r-- 1 root root 3.7K Jan 5 12:00 output_config.toml
-rw-r--r-- 1 root root 2.4M Jan 5 12:05 output_lef_1d_occupancy.bw
-rw-r--r-- 1 root root 57M Jan 5 12:05 output.cool
-rw-r--r-- 1 root root 14K Jan 5 12:05 output.logTroubleshooting problems inside the container is a bit tedious. Any tips?
Yes! For troubleshooting we recommend using an interactive shell running inside the container.
docker run -v "$(pwd -P)/mydata/:/input_data/:ro" \
-v "$(pwd -P)/output/:/output_data/" \
-it --rm --entrypoint=/bin/bash \
ghcr.io/paulsengroup/modle:1.1.0This will drop you in an interactive bash shell.
Press Ctrl+C, Ctrl+D or type exit to leave the shell and stop the container.
root@25e8efe74294:/data# ls -lah
total 4.0K
drwxr-xr-x 2 root root 2 Jan 5 12:00 .
drwxr-xr-x 20 root root 27 Jan 5 12:00 ..
root@25e8efe74294:/data# cd /input_data/
root@25e8efe74294:/input_data# ls -lah
total 250K
drwx------ 2 1000 1000 80 Jan 5 12:00 .
drwxr-xr-x 20 root root 27 Jan 5 12:00 ..
-rw------- 1 1000 1000 365 Jan 5 12:00 grch38.chrom.sizes
-rw------- 1 1000 1000 241K Jan 5 12:00 grch38_h1_extrusion_barriers.bed.xz
root@25e8efe74294:/input_data# touch foo
touch: cannot touch 'foo': Read-only file system
root@25e8efe74294:/input_data# cd /output_data/
root@25e8efe74294:/output_data# ls -lah
total 2.0K
drwxr-xr-x 2 1000 1000 40 Jan 5 12:00 .
drwxr-xr-x 20 root root 27 Jan 5 12:00 ..
root@25e8efe74294:/output_data# touch foo
root@25e8efe74294:/output_data# ls -lah
total 2.0K
drwxr-xr-x 2 1000 1000 60 Jan 5 12:00 .
drwxr-xr-x 20 root root 27 Jan 5 12:00 ..
-rw-r--r-- 1 root root 0 Jan 5 12:01 foo
root@25e8efe74294:/output_data# whereis modle
modle: /usr/local/bin/modle
root@25e8efe74294:/output_data# modle --help
High-performance stochastic modeling of DNA loop extrusion interactions.
Usage: modle [OPTIONS] SUBCOMMAND
Options:
-h,--help Print this help message and exit
-V,--version Display program version information and exit
--config Path to MoDLE's config file (optional).
Subcommands:
simulate, sim Simulate loop extrusion and write resulting molecular contacts in a .cool file.
root@25e8efe74294:/output_data# exit
exitFor Singularity/Apptainer this is even easier:
singularity shell docker://ghcr.io/paulsengroup/modle:1.1.0MoDLE package for Linux and MacOS is available on bioconda and can be installed as follows:
user@dev:/tmp$ conda create -n modle -c conda-forge -c bioconda modle
(modle) user@dev:/tmp$ conda activate modle
(modle) user@dev:/tmp$ whereis modle
modle: /home/user/.miniconda3/envs/modle/bin/modle
(modle) user@dev:/tmp$ modle --version
MoDLE-v1.1.0-biocondaDownload file modle-1.1.0-x86_64-linux.tar.xz from the latest release.
Extract the archive and copy modle and/or modle_tools to a location in your PATH
click to expand
user@dev:/tmp$ curl -LO 'https://github.com/paulsengroup/modle/releases/download/v1.1.0/modle-1.1.0-x86_64-linux.tar.xz'
user@dev:/tmp$ tar -xvf modle-1.1.0-x86_64-linux.tar.xz
modle-1.1.0-x86_64-linux/
modle-1.1.0-x86_64-linux/share/
modle-1.1.0-x86_64-linux/share/licenses/
modle-1.1.0-x86_64-linux/share/licenses/modle_tools/
modle-1.1.0-x86_64-linux/share/licenses/modle_tools/LICENSE
modle-1.1.0-x86_64-linux/share/licenses/modle/
modle-1.1.0-x86_64-linux/share/licenses/modle/LICENSE
modle-1.1.0-x86_64-linux/bin/
modle-1.1.0-x86_64-linux/bin/modle_tools
modle-1.1.0-x86_64-linux/bin/modle
user@dev:/tmp modle-1.1.0-x86_64-linux/bin/modle --version
MoDLE-v1.1.0
# Optional: add modle and modle_tools to your PATH (please ensure $HOME/.local/bin/ is in your PATH)
user@dev:/tmp$ install -Dm0755 modle-1.1.0-x86_64-linux/bin/modle "$HOME/.local/bin/"
user@dev:/tmp$ install -Dm0755 modle-1.1.0-x86_64-linux/bin/modle_tools "$HOME/.local/bin/"
user@dev:/tmp$ whereis modle
modle: /home/user/.local/bin/modle
user@dev:/tmp$ modle --version
MoDLE-v1.1.0MoDLE can be compiled on most UNIX-like systems, including many Linux distributions and MacOS (10.15+).
Here we assume MoDLE was correctly installed and is available in your PATH (i.e. running whereis modle prints something like modle: /usr/local/bin/modle).
If you are using Docker or Singularity/Apptainer, please make sure input files can be reached from within the container (see troubleshooting steps from Using MoDLE with Docker or Singularity/Apptainer.
Folder examples/data/ from this repository contains the input files used throughout this section.
Data provenance
examples/data/hg38.chrom.sizeswas generated from the hg38.chrom.sizes released by UCSC. Chromosmes were filtered and sorted usinggrep -E 'chr[0-9XY]+[[:space:]]' hg38.chrom.sizes | sort -Vexamples/data/hg38_extrusion_barriers.bed.xzwas generated as described the Methods of MoDLE's paper (third paragraph of section "MoDLE simulations"). Refer to code hosted on github.com/paulsengroup/2021-modle-paper-001-data-analysis for more details (useful links: link1, link2, link3, link4. Feel free to get in touch if you are struggling to navigate code hosted in the data analysis repository).
Running a simulation with default settings only requires two input files:
- A chrom.sizes file with the list of chromosomes to be simulated
- A BED file with the list of extrusion barriers to include as part of the simulation
Input files using common compression formats (namely Bzip2, Gzip, LZ4, xz/LZMA and zstd) are supported.
The extrusion barrier BED file should have at least the first 6 columns defined (i.e. chrom, start, end, name, score and strand. See bedtools docs for more details).
Some remarks:
-
chrom should contain the name of one of the chromosome defined in the
.chrom.sizes -
The start and end fields are used to position barriers along chromosomes. MoDLE models extrusion barriers as 1bp entities. Thus, when
$end - start \gt 1$ the extrusion barrier will be placed in the middle of interval$[start, end)$ . -
The name field is required but ignored, and can be safely set to
. -
score is required and should be set to a number between 0 and 1. This number expresses the average occupancy of the extrusion barrier site defined by the current line. Occupancies between 0.7-0.9 can be used as a starting point.
Check out this tutorial to learn how barrier occupancy can be inferred from CTCF/RAD21 ChIP-Seq data. More information regarding how extrusion barriers are modeled canm be found in MoDLE's paper and suppl. text.
-
strand is required and is used to define the extrusion barrier direction (should be one of
-,+or.). As of MoDLE v1.1.0, extrusion barriers are modeled after CTCF barriers. Thus, the strand field should be populated with the direction of the CTCF binding site that is being defined. Barriers without strand information (i.e. with strand.) are ignored.
user@dev:/tmp/modle$ modle simulate \
--chrom-sizes examples/data/hg38.chrom.sizes \
--extrusion-barrier-file examples/data/hg38_extrusion_barriers.bed.xz \
--output-prefix examples/out/hg38_default
[2023-05-11 11:47:15.718] [info]: running MoDLE v1.1.0
[2023-05-11 11:47:15.718] [info]: complete log will be written to file "examples/out/hg38_default.log"
[2023-05-11 11:47:15.718] [info]: writing simulation parameters to config file "examples/out/hg38_default_config.toml"
[2023-05-11 11:47:15.718] [info]: command: modle simulate --chrom-sizes examples/data/hg38.chrom.sizes --extrusion-barrier-file examples/data/hg38_extrusion_barriers.bed.xz --output-prefix examples/out/hg38_default
[2023-05-11 11:47:15.719] [info]: simulation will use up to 16 out of 16 available CPU cores.
[2023-05-11 11:47:15.719] [info]: using --target-contact-density=1.00 as stopping criterion.
[2023-05-11 11:47:15.719] [info]: contact sampling strategy: tad-plus-loop-with-noise.
[2023-05-11 11:47:15.719] [info]: contact matrix resolution: 5000bp
[2023-05-11 11:47:15.719] [info]: importing chromosomes from "examples/data/hg38.chrom.sizes"...
[2023-05-11 11:47:15.719] [info]: imported 24 chromosomes in 239.985us.
[2023-05-11 11:47:15.719] [info]: importing extrusion barriers from "examples/data/hg38_extrusion_barriers.bed.xz"...
[2023-05-11 11:47:15.761] [info]: imported 38815 barriers from "examples/data/hg38_extrusion_barriers.bed.xz" in 41.776052ms.
[2023-05-11 11:47:15.762] [info]: spawning worker thread W0...
[2023-05-11 11:47:15.762] [info]: spawning worker thread W1...
[2023-05-11 11:47:15.762] [info]: spawning worker thread W3...
...
[2023-05-11 11:47:15.764] [info]: begin processing chr1:0-248956422: simulating ~37485 epochs across 512 cells using 4979 LEFs and 3518 barriers (~73 epochs per cell)...
[2023-05-11 11:47:37.627] [info]: begin processing chr2:0-242193529: simulating ~37501 epochs across 512 cells using 4844 LEFs and 2974 barriers (~73 epochs per cell)...
[2023-05-11 11:47:43.592] [info]: [io] writing contacts for chr1:0-248956422 to file "examples/out/hg38_default.cool"...
[2023-05-11 11:47:46.182] [info]: [io]: written 29875200 contacts for chr1:0-248956422 to file "examples/out/hg38_default.cool" in 2.590415163s (3.47M nnz out of 29.88M pixels).
[2023-05-11 11:47:46.186] [info]: [io]: writing 1D LEF occupancy profile for chr1:0-248956422 to file "examples/out/hg38_default_lef_1d_occupancy.bw"...
[2023-05-11 11:47:46.196] [info]: [io]: writing 1D LEF occupancy profile for chr1:0-248956422 to file "examples/out/hg38_default_lef_1d_occupancy.bw" took 9.964882ms
[2023-05-11 11:47:57.200] [info]: begin processing chr3:0-198295559: simulating ~37474 epochs across 512 cells using 3966 LEFs and 2469 barriers (~73 epochs per cell)...
...
[2023-05-11 11:51:49.400] [info]: [io] writing contacts for chrY:0-57227415 to file "examples/out/hg38_default.cool"...
[2023-05-11 11:51:49.744] [info]: [io]: written 6867600 contacts for chrY:0-57227415 to file "examples/out/hg38_default.cool" in 344.473166ms (0.92M nnz out of 6.87M pixels).
[2023-05-11 11:51:49.744] [info]: [io]: writing 1D LEF occupancy profile for chrY:0-57227415 to file "examples/out/hg38_default_lef_1d_occupancy.bw"...
[2023-05-11 11:51:49.746] [info]: [io]: writing 1D LEF occupancy profile for chrY:0-57227415 to file "examples/out/hg38_default_lef_1d_occupancy.bw" took 1.565303ms
[2023-05-11 11:51:51.925] [info]: simulation terminated without errors in 4m36.206746164s!
Bye.The above command will create folder examples/out/ (if it doesn't already exist), and write the following files inside it:
examples/out
├── hg38_default_config.toml
├── hg38_default.cool
├── hg38_default_lef_1d_occupancy.bw
└── hg38_default.log
Output files produced by the above command are available here.
Simulated interactions are written to file hg38_default.cool. More information about the .cool format is available at open2c/cooler.
In case any of the output files already exist, MoDLE will refuse to run and print an error message listing the file name collisions.
Passing the --force flag overrides this behavior and will cause MoDLE to overwrite existing files.
To visualize .cool files we recommend using cooler show or higlass (more specifically higlass-docker or higlass-manage).
Tips for visualization
Quickly visualizing interactions with Cooler
cooler show examples/out/hg38_default.cool chr1:5000000-8000000
Visualizing interactions with HiGlass
Before visualizing heatmaps with HiGlass, the single-resolution .cool file produced by MoDLE needs to be converted to a multi-resolution Cooler (.mcool) file.
This is easily done with cooler zoomify:
user@dev:/tmp/modle$ cooler zoomify examples/out/hg38_default.cool
INFO:cooler.cli.zoomify:Recursively aggregating "examples/out/hg38_default.cool"
INFO:cooler.cli.zoomify:Writing to "examples/out/hg38_default.mcool"
INFO:cooler.reduce:Copying base matrices and producing 12 new zoom levels.
INFO:cooler.reduce:Bin size: 5000
INFO:cooler.reduce:Aggregating from 5000 to 10000.
INFO:cooler.create:Creating cooler at "examples/out/hg38_default.mcool::/resolutions/10000"
INFO:cooler.create:Writing chroms
INFO:cooler.create:Writing bins
INFO:cooler.create:Writing pixels
INFO:cooler.reduce:0 10000066
INFO:cooler.reduce:10000066 20000079
INFO:cooler.reduce:20000079 30000056
INFO:cooler.reduce:30000056 40000018
INFO:cooler.reduce:40000018 43586992
INFO:cooler.create:Writing indexes
INFO:cooler.create:Writing info
INFO:cooler.reduce:Aggregating from 10000 to 20000.
...Next, you need to start HiGlass server.
If you are using higlass-manage, you may have to prefix commands with sudo or sudo -E.
# Using higlass-docker
user@dev:/tmp/modle$ sudo docker pull higlass/higlass-docker
Using default tag: latest
latest: Pulling from higlass/higlass-docker
Digest: sha256:44086069ee7d4d3f6f3f0012569789ec138f42b84aa44357826c0b6753eb28de
Status: Image is up to date for higlass/higlass-docker:latest
docker.io/higlass/higlass-docker:latest
user@dev:/tmp/modle$ sudo docker run \
--detach \
--publish 8989:80 \
--volume ~/hg-data:/data \
--volume ~/hg-tmp:/tmp \
--name higlass-modle \
higlass/higlass-docker
eb3fe07ea23b57eb844a590e7f1e939cf82a11e5d25db87c4b25024a41c9a546
# Using higlass-manage
user@dev:/tmp/modle$ sudo higlass-manage start --use-redis --no-public-data
Stopping previously running container
Attempting to remove existing Docker network instance
Stopping previously running Redis container
Pulling redis:5.0.3-alpine
done
Pulling latest image...
done
Data directory: /home/user/hg-data
Temp directory: ()
Starting... default 8989
Docker started: higlass-manage-container-default
sending request 1
Waiting to start (tilesets)...
sending request 2
...
public_data: False
ret: b'{"uid": "default_local"}'
ret: ExecResult(exit_code=0, output=b'')
Replaced js file
StartedNow if you visit localhost:8989/app with your browser you should see an empty HiGlass page like the following:

Next, we need to ingest our .mcool into HiGlass:
user@dev:/tmp/modle$ mkdir ~/hg-data/media/modle
user@dev:/tmp/modle$ cp examples/out/hg38_default.mcool ~/hg-data/media/modle
# With higlass-docker
user@dev:/tmp/modle$ sudo docker exec higlass-modle \
python higlass-server/manage.py ingest_tileset \
--project-name "modle" \
--filename modle/hg38_default.mcool \
--filetype cooler \
--datatype matrix \
--no-upload
# With higlass-manage
user@dev:/tmp/modle$ sudo higlass-manage ingest --project-name=modle modle/hg38_default.mcool --no-upload
state True
Inferred filetype: cooler
Inferred datatype: matrix
state True
name_text:
hg_name: default
command: python higlass-server/manage.py ingest_tileset --filename modle/hg38_default.mcool --filetype cooler --datatype matrix --project-name "modle" --no-upload --uid emFfbA9tTCiBTt6b5vTG8QFinally, after refreshing page localhost:8989/app you should be able to add tileset hg38_default.mcool to the center panel:

In case you are struggling to visualize MoDLE's output feel free to reach out by starting a new discussion.
Tips for programmatic access to Cooler files
Programmatic access to contacts stored in Cooler files is possible using the cooler CLI interface or Python API.
CLI interface
# Dumping interactions in BEDPE format
user@dev:/tmp/modle$ cooler dump --table pixels --join examples/out/hg38_default.cool | head
chr1 0 5000 chr1 0 5000 9
chr1 0 5000 chr1 5000 10000 5
chr1 0 5000 chr1 10000 15000 2
chr1 0 5000 chr1 15000 20000 2
chr1 0 5000 chr1 20000 25000 1
chr1 0 5000 chr1 25000 30000 2
chr1 0 5000 chr1 30000 35000 3
chr1 0 5000 chr1 35000 40000 1
chr1 0 5000 chr1 40000 45000 2
chr1 0 5000 chr1 45000 50000 1
user@dev:/tmp/modle$ cooler dump --table pixels --join examples/out/hg38_default.mcool::/resolutions/200000 | head
chr1 0 20000 chr1 0 20000 114
chr1 0 20000 chr1 20000 40000 120
chr1 0 20000 chr1 40000 60000 69
chr1 0 20000 chr1 60000 80000 59
chr1 0 20000 chr1 80000 100000 34
chr1 0 20000 chr1 100000 120000 28
chr1 0 20000 chr1 120000 140000 25
chr1 0 20000 chr1 140000 160000 18
chr1 0 20000 chr1 160000 180000 20
chr1 0 20000 chr1 180000 200000 12Python API
In [1]: import cooler
In [2]: c = cooler.Cooler("examples/out/hg38_default.cool")
In [3]: selector = c.matrix(balance=False)
In [4]: selector.fetch("chr1:0-50000")
Out[4]:
array([[ 9, 5, 2, 2, 1, 2, 3, 1, 2, 1],
[ 5, 9, 9, 15, 3, 3, 8, 1, 8, 1],
[ 2, 9, 12, 26, 11, 7, 3, 8, 5, 2],
[ 2, 15, 26, 25, 27, 16, 12, 14, 10, 7],
[ 1, 3, 11, 27, 20, 42, 30, 17, 15, 10],
[ 2, 3, 7, 16, 42, 21, 27, 17, 15, 12],
[ 3, 8, 3, 12, 30, 27, 26, 27, 26, 17],
[ 1, 1, 8, 14, 17, 17, 27, 23, 44, 23],
[ 2, 8, 5, 10, 15, 15, 26, 44, 31, 35],
[ 1, 1, 2, 7, 10, 12, 17, 23, 35, 23]], dtype=int32)
In [5]: selector = c.matrix(balance=False, as_pixels=True, join=True)
In [6]: selector.fetch("chr1:0-50000")
Out[6]:
chrom1 start1 end1 chrom2 start2 end2 count
0 chr1 0 5000 chr1 0 5000 9
1 chr1 0 5000 chr1 5000 10000 5
2 chr1 0 5000 chr1 10000 15000 2
3 chr1 0 5000 chr1 15000 20000 2
4 chr1 0 5000 chr1 20000 25000 1
5 chr1 0 5000 chr1 25000 30000 2
6 chr1 0 5000 chr1 30000 35000 3
7 chr1 0 5000 chr1 35000 40000 1
8 chr1 0 5000 chr1 40000 45000 2
9 chr1 0 5000 chr1 45000 50000 1
...Refer to Cooler documentation for more details.
The hg38_default_config.toml file produced by MoDLE in the previous section is a config file that can be used to repeat simulations using the same set of parameters and input files.
Information found in config files is also embedded in the .cool file as a JSON string, and can be extracted using cooler info:
user@dev:/tmp/modle$ cooler info --metadata examples/out/hg38_default.cool
{
"simulate": {
"avg-lef-processivity": 300000,
"burnin-extr-speed-coefficient": 1,
"burnin-history-length": 100,
"burnin-smoothing-window-size": 5,
"burnin-target-epochs-for-lef-activation": 320,
"chrom-sizes": "examples/data/hg38.chrom.sizes",
...The following will run a simulation with the same settings as the simulation from the previous example
modle simulate --config examples/out/hg38_default_config.toml --forceParameters read from a config file have lower priority than parameter specified through the CLI, meaning that CLI options can be used to override parameters read from the config file.
This can be useful when running a batch of simulations using a large number of optional parameters, and where only a handful of parameters need to change across simulation runs.
The following command will run a simulation using parameters from the previous example as starting point, but using a different number of cells and target contact density.
modle simulate \
--config examples/out/hg38_default_config.toml \
--ncells 16 \
--target-contact-density 10 \
--output-prefix examples/out/hg38_deepOutput files produced by the above command are available here.

MoDLE's config files are in TOML format.
Adding a line like my-option=my_value to the config file has the same effect as passing --my-option=my_value on the CLI.
For an up-to-date list of supported CLI options, please refer to MoDLE's help message:
user@dev:/tmp/modle$ modle simulate --help
Simulate loop extrusion and write resulting molecular contacts in a .cool file.
Usage: modle simulate [OPTIONS]
Options:
-h,--help Print this help message and exit
[Option Group: IO]
Options controlling MoDLE input, output and logs.
Options:
-c,--chrom-sizes REQUIRED Path to file with chromosome sizes in chrom.sizes format.
-b,--extrusion-barrier-file REQUIRED
Path to a file in BED6+ format with the genomic coordinates of extrusion barriers to be
simulated. The score field in a BED record should be a number between 0 and 1 and is
interpreted as the extrusion barrier occupancy for the extrusion barrier described
by the record.
Barriers mapping on chromosomes not listed in the chrom.sizes file passed through
the --chrom-sizes option are ignored.
-g,--genomic-intervals Path to BED3+ file with the genomic regions to be simulated.
Intervals listed in this file should be a subset of the chromosomes defined in the .chrom.sizes files.
Intervals referring to chromosomes not listed in the .chrom.sizes file are ignored.
-f,--force Overwrite existing files (if any).
-o,--output-prefix REQUIRED Output prefix.
Can be an absolute or relative path including the file name but without the extension.
Example: running modle sim -o /tmp/my_simulation ... yields the following files:
- /tmp/my_simulation.cool
- /tmp/my_simulation.log
- /tmp/my_simulation_config.toml
- /tmp/my_simulation_lef_1d_occupancy.bw
--assembly-name=unknown Name of the genome assembly to be simulated.
This is only used to populate the "assembly" attribute in the output .cool file.
...If you use MoDLE in your research, please cite the following paper:
Rossini, R., Kumar, V., Mathelier, A. et al. MoDLE: high-performance stochastic modeling of DNA loop extrusion interactions. Genome Biol 23, 247 (2022). https://doi.org/10.1186/s13059-022-02815-7
BibTex
@article{rossini_2022,
author = {Rossini, Roberto and Kumar, Vipin and Mathelier, Anthony and Rognes, Torbjørn and Paulsen, Jonas},
doi = {10.1186/s13059-022-02815-7},
journal = {Genome Biology},
month = {11},
title = {{MoDLE: high-performance stochastic modeling of DNA loop extrusion interactions}},
url = {https://doi.org/10.1186/s13059-022-02815-7},
volume = {23},
year = {2022}
}In addition to the above reference, you can also cite specific version(s) of MoDLE using one of the DOIs from Zenodo: 10.5281/zenodo.6424697