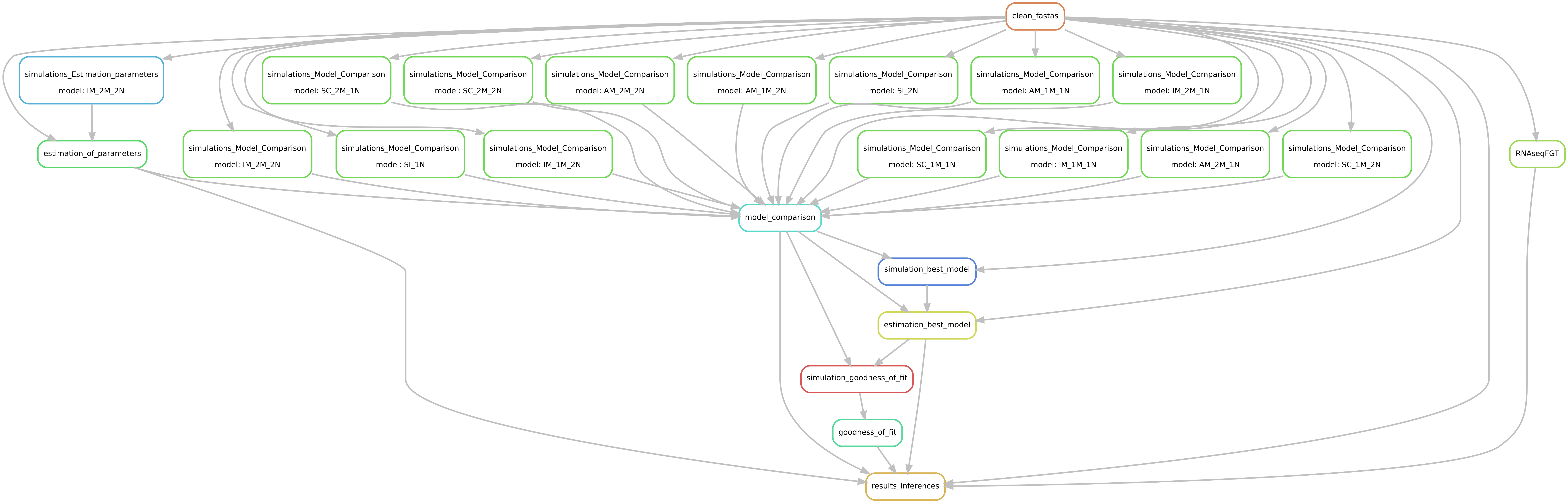
The entire workflow is based on snakemake.
https://snakemake.readthedocs.io/en/stable/
Please adapt the pathway to your system.
snakemake --snakefile /shared/mfs/data/home/croux/softwares/DILS/bin/Snakefile_2pop -p -j 50 --configfile /shared/home/croux/scratch/myProject/config.yaml --cluster-config /shared/home/croux/scratch/myProject/cluster.json --cluster "sbatch --nodes={cluster.node} --ntasks={cluster.n} --cpus-per-task={cluster.cpusPerTask} --time={cluster.time}"
Needs:
1 Snakefile
1 config.yaml file
1 cluster.json file
Executables are all found in the bin/ subdirectory. The pathway of subdirectory has to be indicated in the Snakefiles. This is the only required file modification
bin/fasta2ABC_1pop.py
bin/fasta2ABC_2pops.py
bin/mscalc_1pop_observedDataset_SFS.py
bin/mscalc_1pop_SFS.py
bin/mscalc_2pop_observedDataset.py
bin/mscalc_2pop_observedDataset_SFS.py
bin/mscalc_2pop.py
bin/mscalc_2pop_SFS.py
bin/priorgen_1pop.py
bin/priorgen_2pop_popGrowth.py
bin/priorgen_2pop.py
bin/priorgen_gof_1pop.py
bin/priorgen_gof_2pop_popGrowth.py
bin/priorgen_gof_2pop.py
bin/priorgen_gof_2pop_test_monolocus.py
bin/submit_simulations_1pop.py
bin/submit_simulations_2pop_popGrowth.py
bin/submit_simulations_2pop.py
bin/submit_simulations_2pop_test_monolocus.py
bin/submit_simulations_gof_1pop.py
bin/submit_simulations_gof_2pop_popGrowth.py
bin/submit_simulations_gof_2pop.py
some scripts uses pypy as python interpreter
from math import ceil
from numpy import log
from numpy import median
from numpy.random import beta
from numpy.random import binomial
from numpy.random import randint
from numpy.random import uniform
from random import choice
from random import randint
from random import sample
from random import shuffle
import os
import random
import sys
import time
uses Rscript from /usr/bin or elsewhere
bin/collaborative_plot.R
bin/estimates_1pop_best.R
bin/estimates_2pop_best.R
bin/estimates_2pop.R
bin/get_parameters_1pop_CV.R
bin/get_parameters_1pop.R
bin/get_parameters_2pop.R
bin/gof_1pop.R
bin/gof_2pop.R
bin/model_comp_1pop_allModels.R
bin/model_comp_2pop_allModels.R
bin/model_comp_2pop_locus.R
bin/model_comp_2pop.R
bin/PCA.R
library(abcrf)
library(data.table)
library(FactoMineR)
library(ggplot2)
library(ggpubr)
library(nnet)
library(plotly)
library(tidyverse)
library(viridis)
Install R libraries for the user interface:
list_libraries = c('shiny', 'shinythemes', 'shinydashboard', 'shinydashboardPlus', 'DT', 'shinyWidgets', 'dashboardthemes', 'devtools', 'shinyhelper', 'plotly', 'viridis', 'tidyr', 'RColorBrewer', 'yaml', 'ggpubr', 'FactoMineR', 'shinycssloaders')
for(lib_tmp in list_libraries){
install.packages(lib_tmp, dep=T)
}
library(devtools)
install_github("nik01010/dashboardthemes")
library(shiny)
library(shinythemes)
library(shinydashboard)
library(shinydashboardPlus)
library(DT)
library(shinyWidgets)
library(dashboardthemes) # library(devtools); install_github("nik01010/dashboardthemes")
library(shinyhelper)
library(plotly)
library(viridis)
library(tidyr)
library(RColorBrewer)
library(yaml)
library(ggpubr)
library(FactoMineR)
library(shinycssloaders)
Can be launched as follows from the webinterface subdirectory:
Rscript app.R host=127.0.0.9 port=8162
C code, compiled by executing the command ./clms (calling gcc) in the msnsam/ directory
C code compiled by: gcc -Wall -o RNAseqFGT RNAseqFGT.c RNAseqFGT_seq_reading.c RNAseqFGT_analysis.c -I RNAseqFGT.h
This file contains informations for Slurm about the submited jobs, in particular, the required resources (CPU, memory, duration).
{
"__default__" :
{
"node" : 1,
"ntasks" : 1,
"n" : 1,
"cpusPerTask" : 1,
"memPerCpu" : 3000,
"time" : "02:00:00"
},
"fasta2ABC_2pops" :
{
"cpusPerTask" : 8,
"time" : "02:00:00",
"memPerCpu" : 2000
},
"RNAseqFGT" :
{
"cpusPerTask" : 1,
"time" : "01:00:00",
"memPerCpu" : 10000
},
"modelComparison" :
{
"cpusPerTask" : 8,
"time" : "01:30:00",
"memPerCpu" : 4000
},
"estimation" :
{
"cpusPerTask" : 8,
"time" : "01:30:00",
"memPerCpu" : 3000
},
"estimation_best_model" :
{
"cpusPerTask" : 8,
"time" : "01:30:00",
"memPerCpu" : 3000
},
"estimation_best_model_2" :
{
"cpusPerTask" : 8,
"time" : "01:30:00",
"memPerCpu" : 3000
},
"estimation_best_model_3" :
{
"cpusPerTask" : 8,
"time" : "01:30:00",
"memPerCpu" : 3000
},
"estimation_best_model_4" :
{
"cpusPerTask" : 8,
"time" : "01:30:00",
"memPerCpu" : 3000
},
"estimation_best_model_5" :
{
"cpusPerTask" : 8,
"time" : "01:30:00",
"memPerCpu" : 3000
},
"locus_modelComp" :
{
"cpusPerTask" : 8,
"time" : "01:30:00",
"memPerCpu" : 3000
},
"PCA_SS" :
{
"cpusPerTask" : 1,
"time" : "01:00:00",
"memPerCpu" : 5000
}
}
Configuration file used by Snakemake to adapt the workflow to a particular analysis. Contains information such as species names, genomic region (coding, noncoding), prior boundaries, etc...
mail_address: user@gmail.com
infile: /shared/home/croux/scratch/moules/sequences.fas
region: coding
nspecies: 2
nameA: Spring
nameB: Sydney
useSFS: 0
nameOutgroup: NA
config_yaml: /shared/home/croux/scratch/moules/config.yaml
timeStamp: SpringSydney
population_growth: constant
modeBarrier: bimodal
max_N_tolerated: 0.2
Lmin: 100
nMin: 6
mu: 0.00000002763
rho_over_theta: 0.5
N_min: 1000
N_max: 500000
Tsplit_min: 10000
Tsplit_max: 1750000
M_min: 1
M_max: 40