Minicurso - Bioinformática Aplicada à área da Saúde
Ministrado por: Renato Puga
Horário : 09h00 - 15h30
E-mail : renatopuga @ gmail . com
Utilizando a plataforma Galaxy Hub e seguindo o pipeline de chamada de variantes genéticas, nós partimos dos dados brutos (.FASTQ), geramos Relatórios de Qualidade com os programas (FastQC e MultiQC), mapeameamos as sequências no genoma de referência com (BWA-MEM) e finalizamos chamando variantes com o (FreeBayes) até chegarmos ao arquivo de variantes (.VCF).
Referência: [Tutorial NGS] (https://galaxyproject.org/tutorials/ngs/)
- Anderson Santos <andersonsfa0 @ gmail . com> [github L1gth1] (https://github.com/L1ght1)
- Arthur Juan Rabelo Lima <arthurgto.rbelo @ outlook . com> [github arthurjrl] (https://github.com/arthurjrl)
- Késsia Évely Campos Souza <evellycsousa @ gmail . com>
- Leandro Nascimento <leandronasx18 @ gmail . com> [github Leandro-quan] (https://github.com/Leandro-quan)
- Marcos Rios <rioos.marcos @ gmail . com> [marcoslor] (https://github.com/marcoslor)
- Ramon Silva <ramondecerqueirasilva @ gmail . com> [ramondcsilva] (https://github.com/ramondcsilva)
- Wellington Batalha <wsbatalha @ gmail . com>
- Aula Introdução - Clique aqui para acessar os Slides
- Prática com use Galaxy (roteiro abaixo)
Nesta seção, veremos aspectos práticos da manipulação de dados de sequenciamento de próxima geração. Começaremos com o formato FASTQ produzido pela maioria das máquinas de sequenciamento e terminaremos com o formato SAM / BAM representando as leituras mapeadas.
Acesse: [usegalaxy.org] (https://usegalaxy.org/)
Clique no Menu: Login or Register > Register:
Fonte: Galaxy Login or Register. ([usegalaxy] (https://usegalaxy.org))
Copie os links abaixo e siga os passos para carregar os dados na plataforma Galaxy:
https://zenodo.org/record/583613/files/sample1-f.fq.gz
https://zenodo.org/record/583613/files/sample1-r.fq.gz
https://zenodo.org/record/583613/files/sample2-f.fq.gz
https://zenodo.org/record/583613/files/sample2-r.fq.gz
Agora, no Galaxy, clique no ícone  para abrir a janela Download from web or upload from disk , então clique no botão Paste/Fetch data e cole os links das amostras no painel.
para abrir a janela Download from web or upload from disk , então clique no botão Paste/Fetch data e cole os links das amostras no painel.
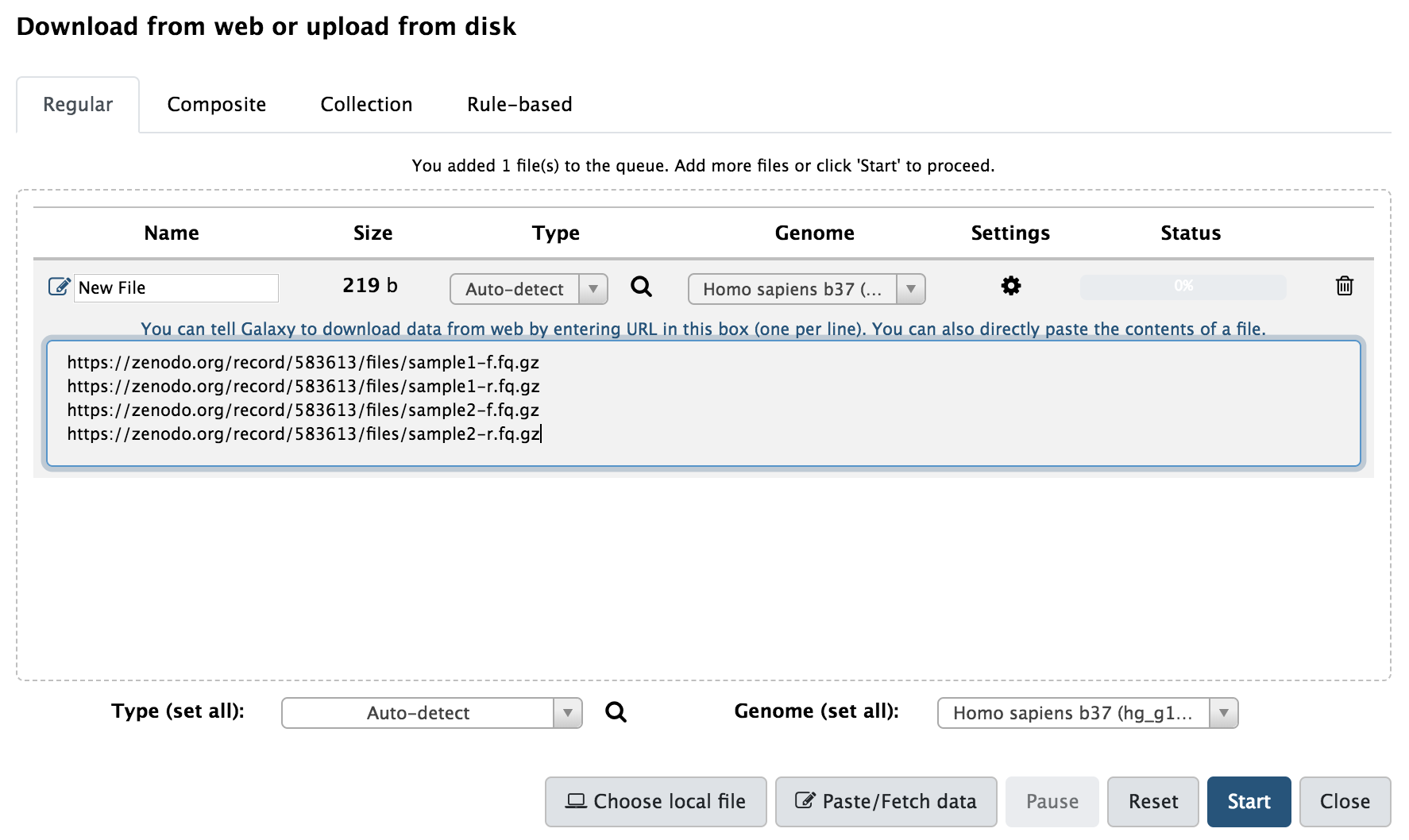
Os dados são paired end data (arquivos com -f são forward e arquivos com -r são reverses) representando duas amostras independentes produzidas por uma máquina Illumina.
O FastQ não é um formato muito bem definido. No início, vários fabricantes de instrumentos de sequenciamento estavam livres para interpretar o FASTQ, resultando em múltipas variações. Essa variação resultou principalmente de diferentes maneiras de codificar os valores de qualidade, conforme descrito aqui (abaixo, você explicará os índices de qualidade e seu significado).
Hoje, a versão FASTQ do Sanger é considerada a forma padrão de FASTQ. O Galaxy está usando o fastq sanger como a única entrada legítima para ferramentas de processamento downstream e fornece vários utilitários para converter arquivos fastq nesse formato (consulte NGS: QC e seção de manipulação de ferramentas do Galaxy).
@M02286:19:000000000-AA549:1:1101:12677:1273 1:N:0:23
CCTACGGGTGGCAGCAGTGAGGAATATTGGTCAATGGACGGAAGTCTGAACCAGCCAAGTAGCGTGCAG
+
ABC8C,:@F:CE8,B-,C,-6-9-C,CE9-CC--C-<-C++,,+;CE<,,CD,CEFC,@E9<FCFCF?9
@M02286:19:000000000-AA549:1:1101:15048:1299 1:N:0:23
CCTACGGGTGGCTGCAGTGAGGAATATTGGACAATGGTCGGAAGACTGATCCAGCCATGCCGCGTGCAG
+
ABC@CC77CFCEG;F9<F89<9--C,CE,--C-6C-,CE:++7:,CF<,CEF,CFGGD8FFCFCFEGCF
@M02286:19:000000000-AA549:1:1101:11116:1322 1:N:0:23
CCTACGGGAGGCAGCAGTAGGGAATCTTCGGCAATGGACGGAAGTCTGACCGAGCAACGCCGCGTGAGT
+
AAC<CCF+@@>CC,C9,F9C9@9-CFFFE@7@:+CC8-C@:7,@EFE,6CF:+8F7EFEEF@EGGGEEE
- Cada sequência é representada por quatro linhas:
- @ sequida pela ID da sequência e informações da corrida são opcionais
- sequência de nucleotído
-
- (separador)
- score de qualidade de cada base codificada com os símbolos da tabela ASCII
Um dos primeiros passos na análise dos dados de NGS é verificar a qualidade dos dados. O FastQC é uma ferramenta que permite avaliar a qualidade dos conjuntos de dados fastq.
Utilize a busca para encontrar o programa FASTQC: NGS: QC and manipulation > FastQC Read Quality reports.
Agora, clique no ícone Multiple datasets e selecione todos os arquivos .fq.gz.
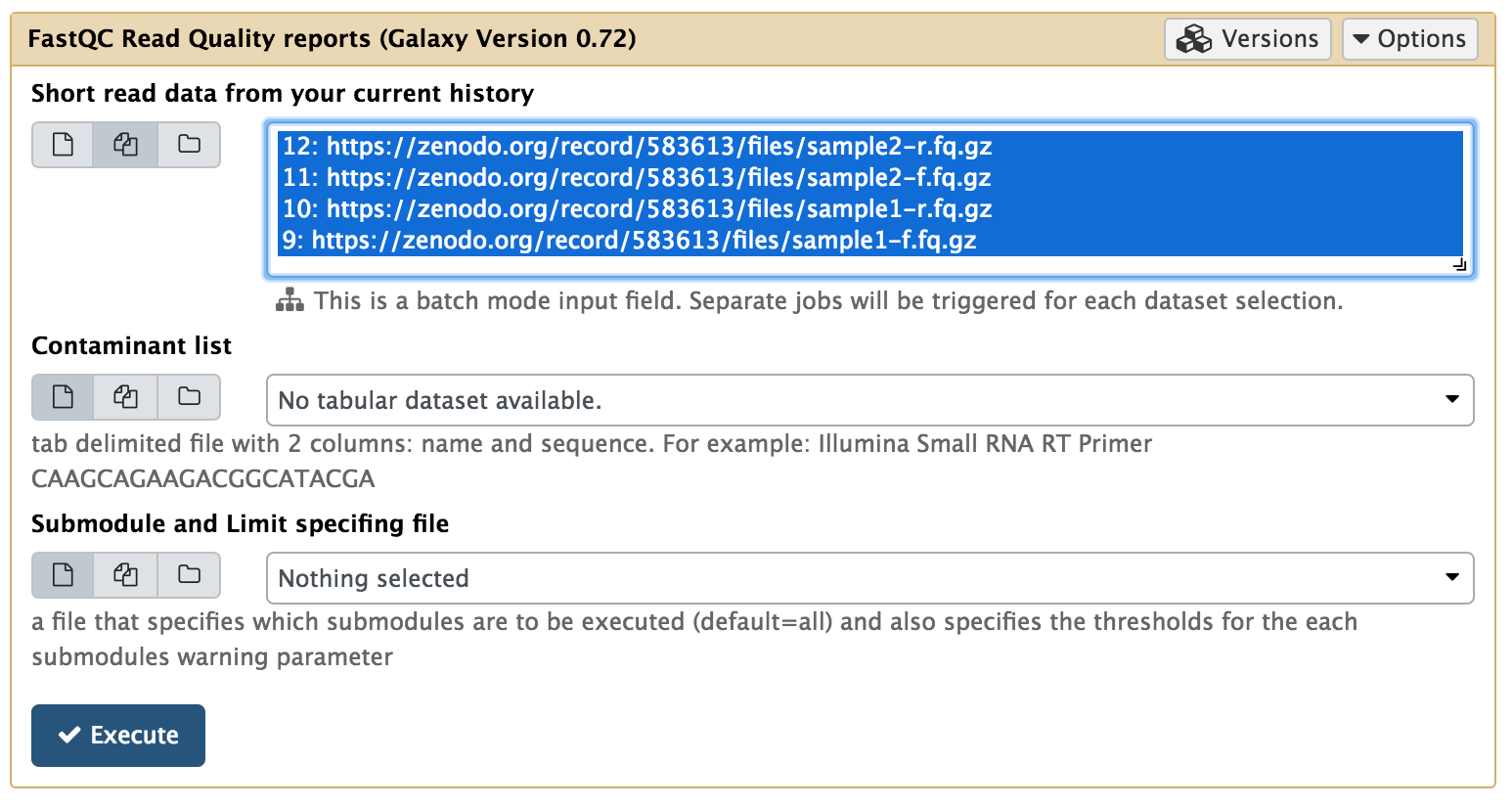
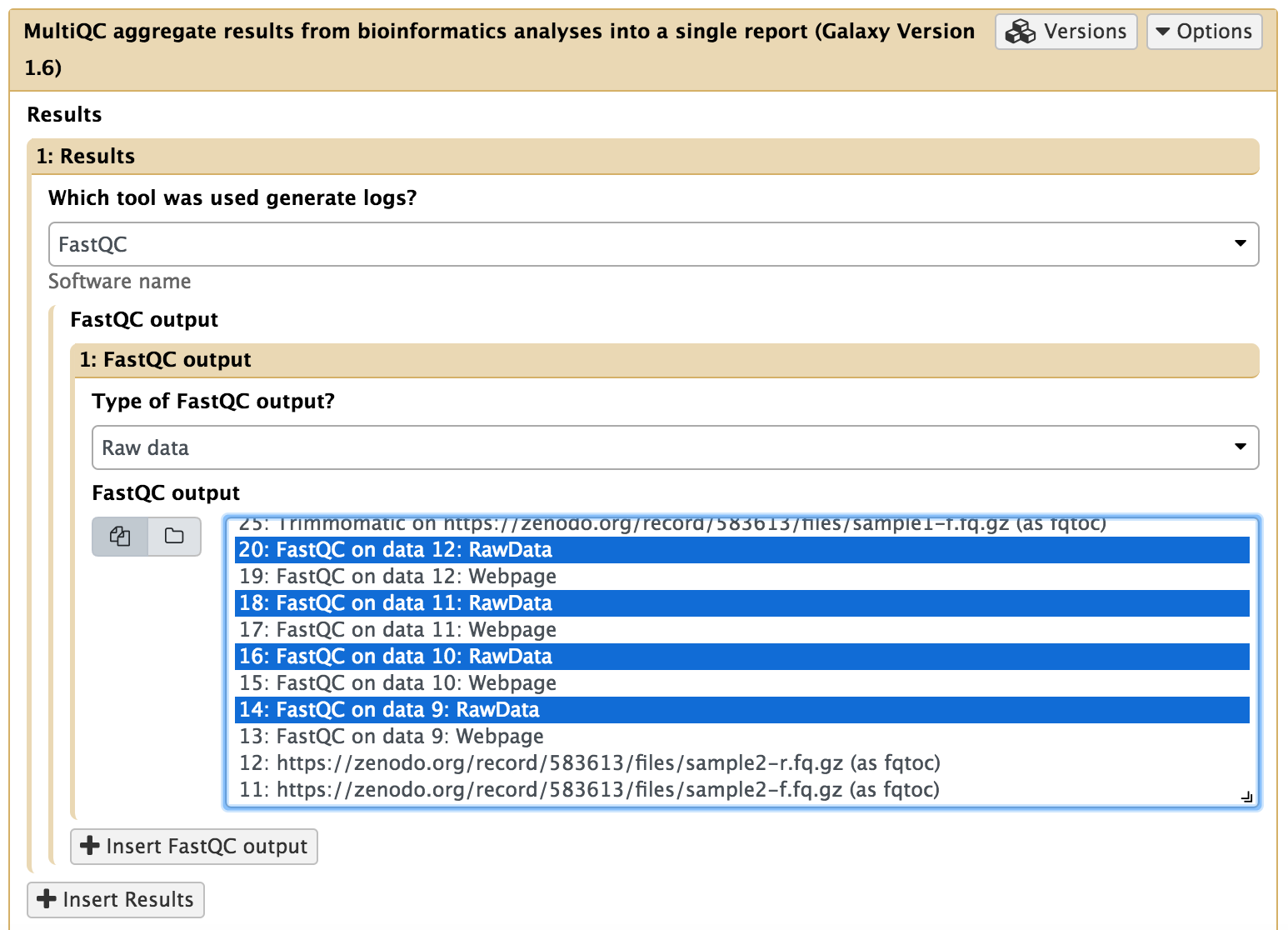
Uma ferramenta modular para agregar resultados de análises de bioinformática em muitas amostras em um único relatório.
Utilize a busca para encontrar o programa MultiQC: NGS: QC and manipulation > MultiQC aggregate results from bioinformatics analyses into a single report.
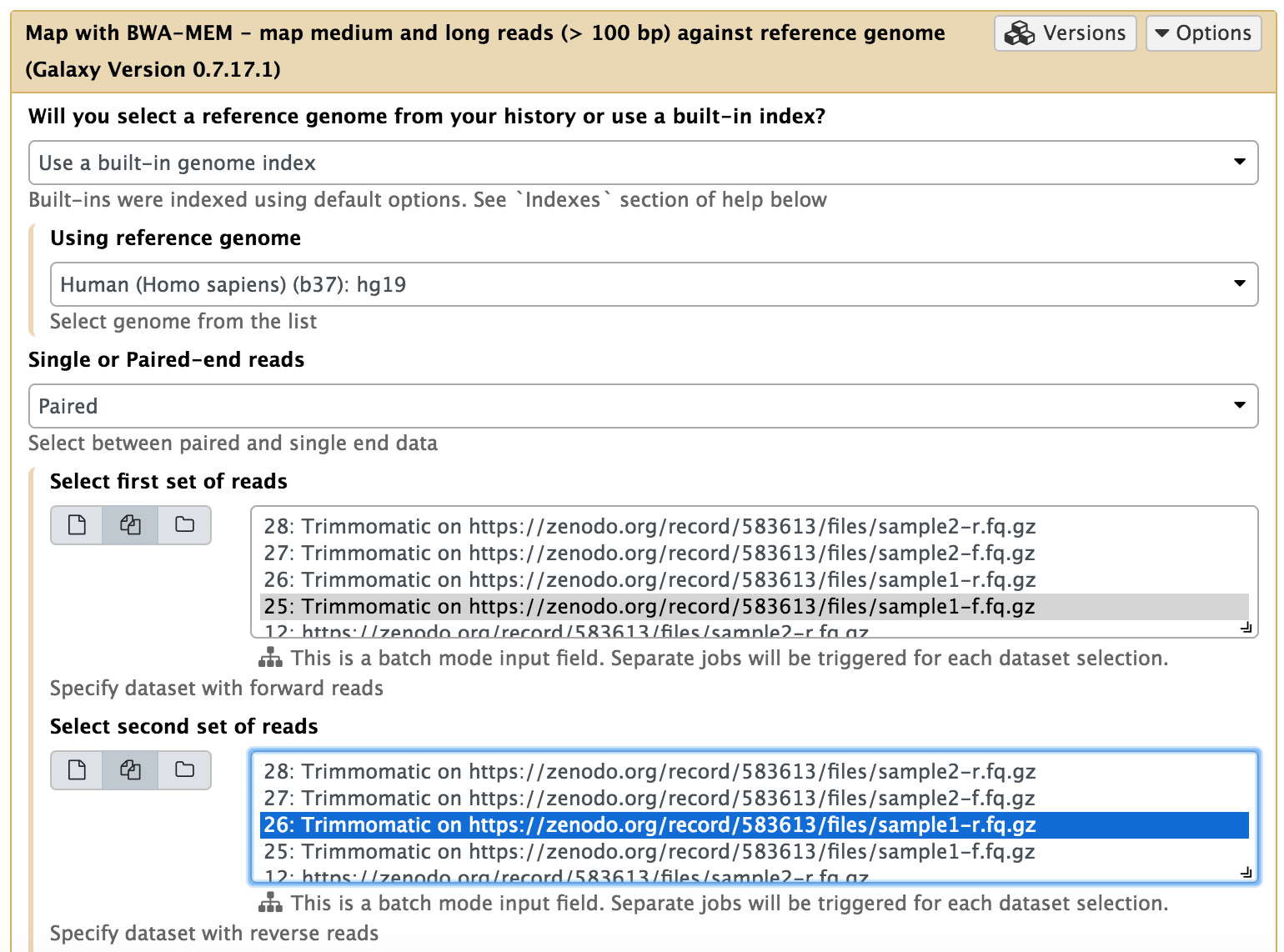
O mapeamento de sequências de NGS contra sequências de referência é uma das principais etapas da análise. Em nosso exemplo vamos utilizar o algorítmo BWA-mem:
- BWA-MEM 2013 - Li
Os mapeadores geralmente comparam as sequências com uma sequência de referência que foi transformada em uma estrutura de dados altamente acessível chamada índice do genoma. Tais índices devem ser gerados antes do início do mapeamento. As instâncias do Galaxy normalmente armazenam índices para várias construções de genoma disponíveis publicamente.
- bwa-mem
- hg19
- paired-end reads
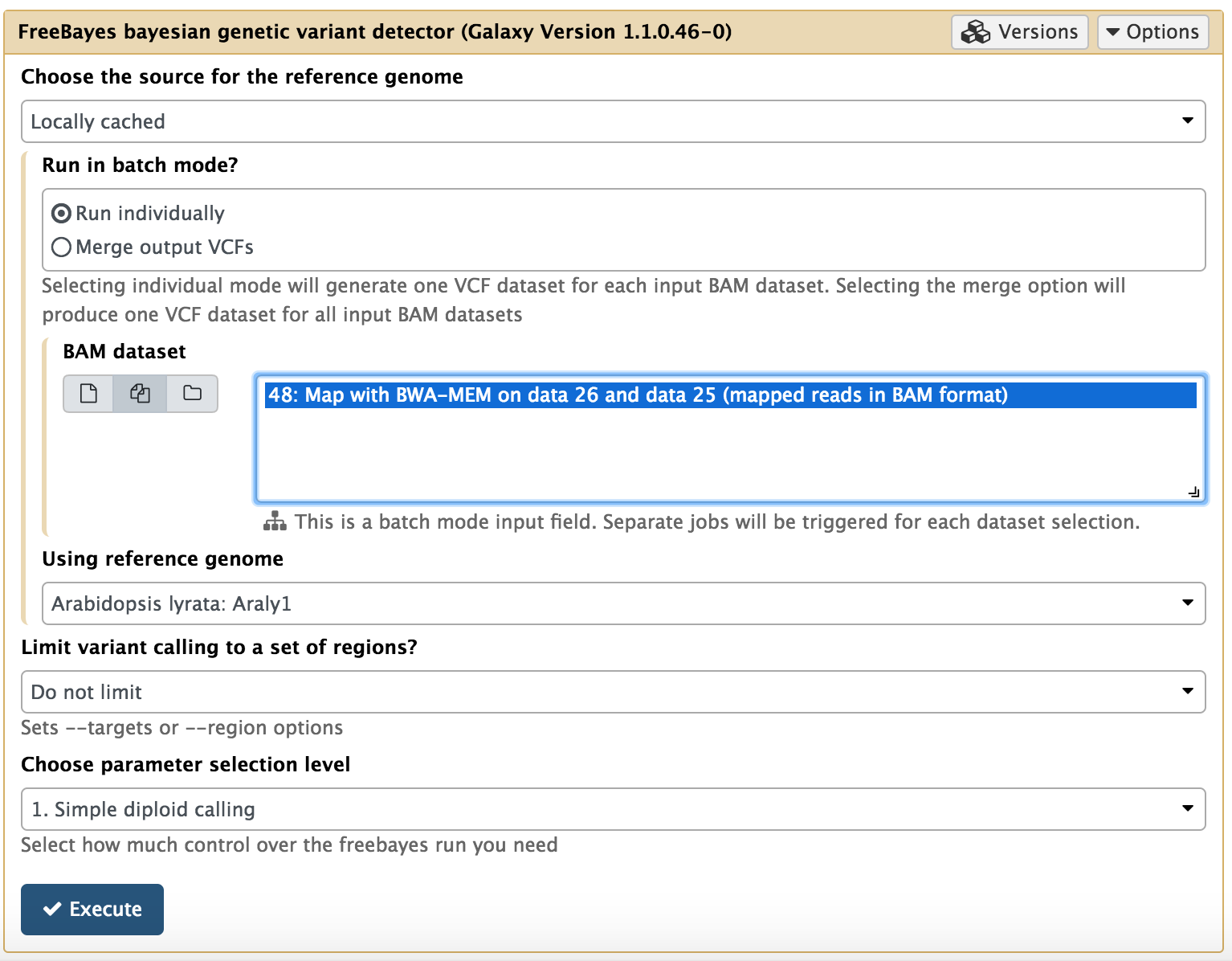
FreeBayes bayesian genetic variant detector. NGS: Variant Analysis > FreeBayes bayesian genetic variant detector