


Master: 

![]()
The Workflow4Metabolomics, W4M in short, is a French infrastructure offering software tool processing, analyzing and annotating metabolomics data. It is based on the Galaxy platform.
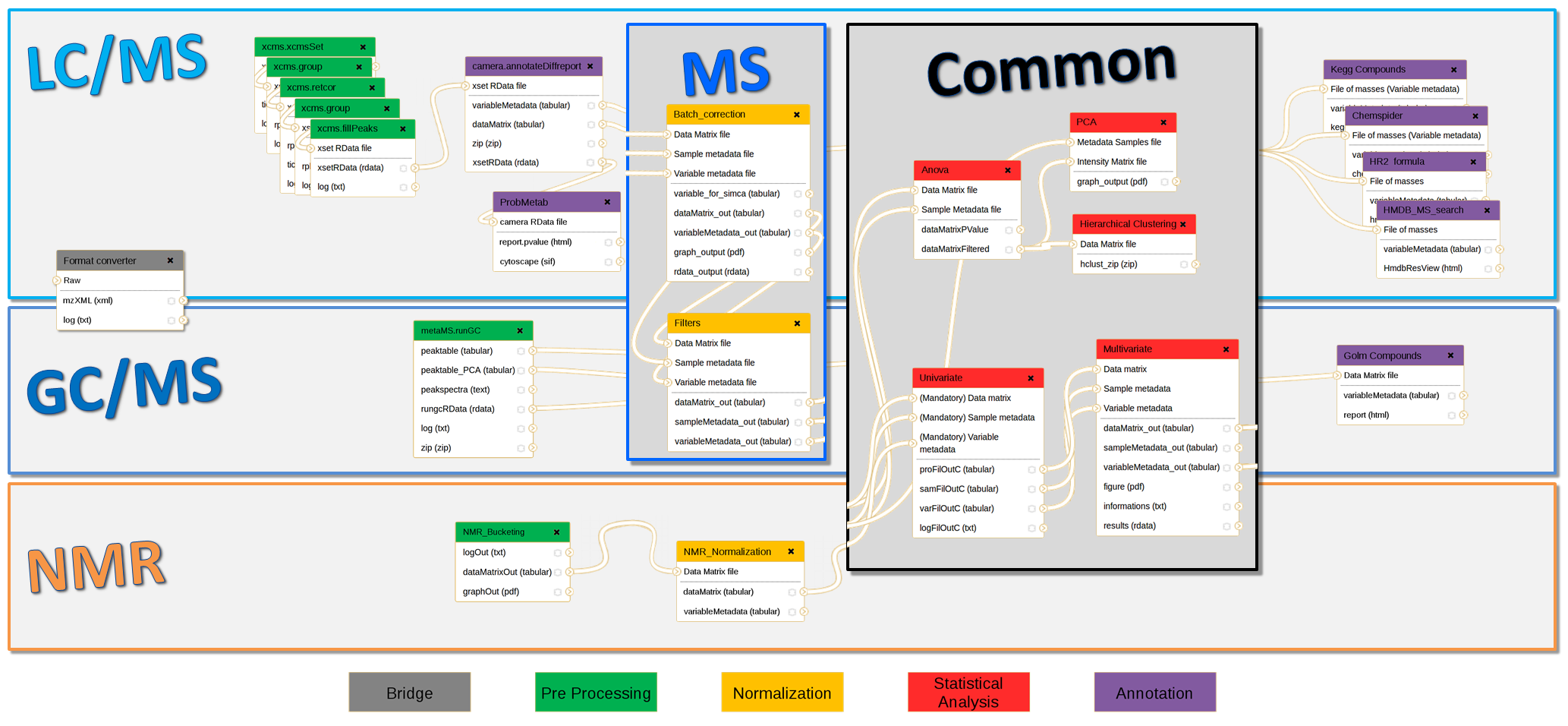
In the context of collaboration between metabolomics (MetaboHUB French infrastructure) and bioinformatics platforms (IFB: Institut Français de Bioinformatique), we have developed full LC/MS, GC/MS and NMR pipelines using Galaxy framework for data analysis including preprocessing, normalization, quality control, statistical analysis and annotation steps. Those modular and extensible workflows are composed with existing components (XCMS and CAMERA packages, etc.) but also a whole suite of complementary homemade tools. This implementation is accessible through a web interface, which guarantees the parameters completeness. The advanced features of Galaxy have made possible the integration of components from different sources and of different types. Thus, an extensible Virtual Research Environment (VRE) is offered to metabolomics communities (platforms, end users, etc.), and enables preconfigured workflows sharing for new users, but also experts in the field.
Giacomoni F., Le Corguillé G., Monsoor M., Landi M., Pericard P., Pétéra M., Duperier C., Tremblay-Franco M., Martin J.-F., Jacob D., Goulitquer S., Thévenot E.A. and Caron C. (2014). Workflow4Metabolomics: A collaborative research infrastructure for computational metabolomics. Bioinformatics, http://dx.doi.org/10.1093/bioinformatics/btu813
Galaxy is an open, web-based platform for data intensive biomedical research. Whether on the free public server or your own instance, you can perform, reproduce, and share complete analyses.
Homepage: https://galaxyproject.org/
This project has for aim to maintain Docker files capable of building a full virtual machine running Galaxy and an instance of the Workflow4Metabolomics.
We are currently pushing our developments on GitHub and on the ToolShed.. Since April of 2016, we also try to prioritize the new system to deal with the dependencies : conda. This work is still in progress. Thus in those VM, you will only find those tools available on the TS, which use the conda dependencies and pass Travis test. Sorry for that!
Conda is package manager that among many other things can be used to manage Python packages. It is becoming the default dependency manager within Galaxy.
This Docker container is based on the quay.io/bgruening/galaxy:16.04 (https://github.com/bgruening/docker-galaxy-stable) as a Galaxy flavour Nested in this Docker image, the script install_tools_wrapper.sh will install tools from ToolSheds using Ansible roles provided by the Galaxy project (https://github.com/galaxyproject/ansible-galaxy-tools) Get more documentations this Docker container on https://github.com/bgruening/docker-galaxy-stable: Usage to save data on this read-only system, using an external Slurm cluster, ...
- Use an existing image available on Docker Hub and start from the Step 1
- From this repository a custom docker image from the Custom Build Section
If you have just build a docker image through the Custom Build Section. Omit workflow4metabolomics/ in the docker image name
From your host:
docker pull workflow4metabolomics/galaxy-workflow4metabolomics # to update the local image version
docker run -d -p 8080:80 workflow4metabolomics/galaxy-workflow4metabolomics
# check that your docker is running
docker ps
# to get a ssh connection and use bash, you need your CONTAINER ID (`docker ps`)
docker exec -i -t ed6031485d06 /bin/bash
Remark: on macOS, since you use docker-machine to run docker, you need to allow port forwarding on your docker-machine instance so the docker container port is forwarded to your host machine. For this you have to run the following command:
docker-machine ssh your_docker_machine_name -f -N -L 8080:localhost:8080From your host:
docker pull workflow4metabolomics/galaxy-workflow4metabolomics # to update the local image version
docker run -i -t -p 8080:80 workflow4metabolomics/galaxy-workflow4metabolomics /bin/bash
From the Docker image:
startup
docker run -d -p 8080:80 -p 8021:21 -p 8022:22 -v /home/user/galaxy_storage/:/export/ workflow4metabolomics/galaxy-workflow4metabolomics
Because Docker container are "read-only", you will lost all your changes within the container at shutdown until you ask the Galaxy instance to write in a folder mounted from the host /home/user/galaxy_storage/ under /export/ into the container.
From a MacOX:
There are some issue with those two features depending on which Docker solution was chosen. See this thread: bgruening/docker-galaxy#210
- Docker for Mac: the export directory works and the SFTP too using Filezilla but not the FTP
- Docker Toolbox: the FTP works but not the export directory
Finally, after maybe a couple of minute (dependeing of your machine), you can connect to the Galaxy portal from a browser running on your host: http://localhost:8080/.
You can login as administrator of your Galaxy instance using the login admin@galaxy.org and the password admin
If you want to add or remove tools from the "official" workflow4metabolomics docker container
git clone --recursive git@github.com:workflow4metabolomics/w4m-docker.git
You can change the tools you want to be installed in w4m-config/tool_list_LCMS.yaml.
From your host:
docker build -t galaxy-workflow4metabolomics .
# check your images
docker images
See Step 1