Parallelization
For impatient readers: How many threads should I use?
RAxML-NG supports three levels of parallelism: CPU instruction (vectorization), intra-node (multithreading) and inter-node (MPI). Unlike with RAxML/ExaML, single RAxML-NG executable can handle all parallelism levels which can be configured in run-time (MPI support is optional and should be enabled at compile-time).
As of v.0.7.0 beta, RAxML-NG only supports fine-grained parallelization across alignment sites. This is the same parallelization approach that has been used in RAxML-PTHREADS and ExaML, and conceptually different from coarse-grained parallelization across tree searches or tree moves as implemented in RAxML-MPI or IQTree-MPI, respectively. With fine-grained parallelization, the number of CPU cores that can be efficiently utilized is limited by the alignment "width" (=number of sites). For instance, using 20 cores on a single-gene protein alignment with 300 sites would be suboptimal, and using 100 cores would most probably result in a huge slowdown. In order to prevent the waste of CPU time and energy, RAxML-NG will warn you -- or, in extreme cases, even refuse to run -- if you try to assign too few alignment sites per core.
If you want to utilize many CPU cores for analyzing a small ("single-gene") alignment, please have a look at our ParGenes pipeline which implements coarse-grained parallelization and dynamic load balancing. Alternatively, coarse-grained parallelization can be easily emulated by starting multiple RAxML-NG instances with distinct random seeds. For instance, one can infer 1000 bootstrap trees by calling RAxML-NG five times with --bs-trees 200 (e.g., on five distinct compute nodes). Then, all resulting *.raxml.bootstraps files can be simply concatenated. TODO: tutorial link
By default, RAxML-NG will start as many threads as there are CPU cores available in your system. Most modern CPUs employ so-called hyperthreading technology, which makes each physical core appear as two logical cores to software. For instance, on my laptop with Intel i7-3520M processor, RAxML-NG will detect 4 (logical) cores and use 4 threads by default, even though this CPU has only 2 physical cores. Hyperthreading can be beneficial for some programs, but RAxML-NG shows best performance with one thread per physical core. Thus, I would recommend using the --threads option to set the number of threads manually, to be on the safe side.
If compiled with MPI support, RAxML-NG can leverage multiple compute nodes for a single analysis. Please check your cluster documentation for system-specific instructions for running MPI programs. In MPI-only mode, you should start 1 MPI process per physical CPU core (the number of threads will be set to 1 by default). However, in most cases, hybrid MPI/pthreads setup will be more efficient in terms of both runtime and memory consumption. Typically, you would start 1 MPI rank per compute node, and 1 thread per physical core (e.g. --threads 16 for nodes equipped with dual-slot octa-core CPUs). Once again, please consult your cluster documentation to find out how to properly configure a hybrid MPI/pthreads run. Please note that incorrect configuration can result in extreme slowdowns und hence waste of time and resources!
For attaining optimal performance, it is crucial to ensure that only one RAxML-NG thread is running on each physical CPU core. Usually, operating system can handle thread-to-core assignment automatically. However, some (misconfigured) MPI runtimes tend to pack all threads on a single CPU core, resulting in abysmal performance. To avoid this situation, each thread can be "pinned" to a particular CPU core.
In RAxML-NG, thread pinning is enabled by default only in hybrid MPI/pthreads mode when 1 MPI rank per node is used.
You can explicitly enable or disable thread pinning with --extra thread-pin and --extra thread-nopin, respectively.
RAxML-NG will automatically detect the best set of vector instructions available on your CPU, and use the respective computational kernels to achieve optimal performance. On modern Intel CPUs, autodetection seems to work pretty well, so most probably you don't need to worry about this. However, you can force RAxML-NG to use a specific set of vector instructions with the --simd option, e.g.
raxml-ng --msa ali.fa --model GTR+G --simd sse
to use SSE3 kernels, or
raxml-ng --msa ali.fa --model GTR+G --simd none
to use non-vectorized (scalar) kernels. This option might be useful for debugging, since distinct vectorized kernel might yield slightly different likelihood values due to numerical reasons.
As always, simple questions are the toughest ones. You might as well ask: How fast should I drive?. In both cases, the answer would be: It depends. Depends on where you drive (dataset), your vehicle (system), and your priorities (time vs. money/energy). In RAxML-NG, we put some warning signs and radar speed guns, for your own safety. As in real life, you are free to ignore them (--force option), which can have two outcomes: (1) earlier arrival, or (2) lost time and money. Fortunately, unlike on the road, you can experiment with RAxML-NG safely, and I encourage you to do so.
A reasonable workflow for analyzing a large dataset would be as follows. First, run RAxML-NG in parser mode, e.g.
raxml-ng --parse --msa ali.fa --model partition.txt
This command will generate a binary MSA file (ali.fa.raxml.rba), which can be loaded by RAxML-NG much faster than the original FASTA alignment. Furthermore, it will print estimated memory requirements and the recommended number of threads for this dataset:
[00:00:00] Reading alignment from file: ali.fa
[00:00:00] Loaded alignment with 436 taxa and 1371 sites
Alignment comprises 1 partitions and 1001 patterns
NOTE: Binary MSA file created: ali.fa.raxml.rba
* Estimated memory requirements : 54 MB
* Recommended number of threads / MPI processes: 4
This recommended number of threads is computed using a simple heuristic and should yield a decent runtime/resource utilization trade-off in most cases. It is also a good starting point for your experimentation: you can now run tree searches with varying number of threads, e.g.
raxml-ng --search --msa ali.fa.raxml.rba --tree rand{1} --seed 1 --threads 2
raxml-ng --search --msa ali.fa.raxml.rba --tree rand{1} --seed 1 --threads 4
raxml-ng --search --msa ali.fa.raxml.rba --tree rand{1} --seed 1 --threads 8
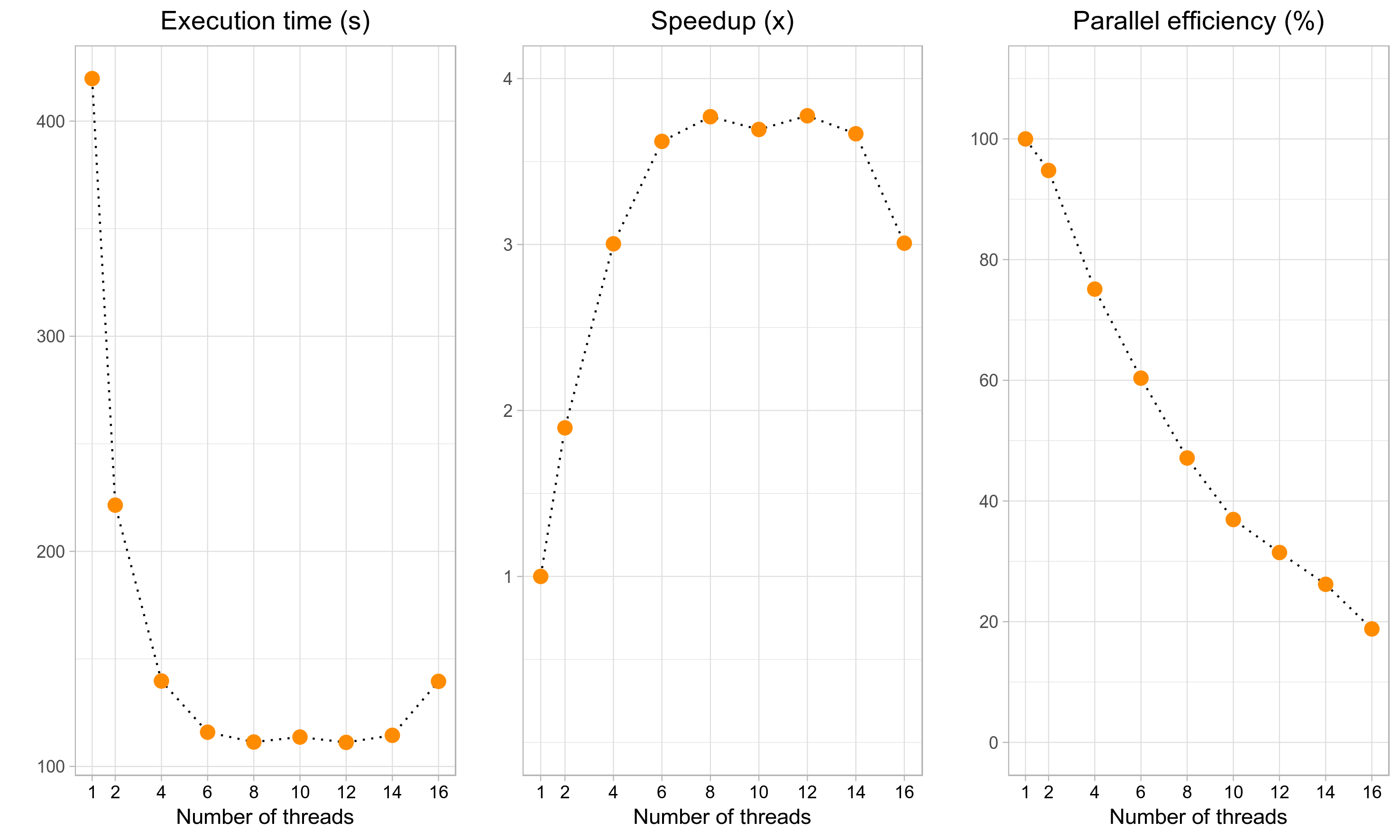
and so on. For a small dataset like in the example above, the execution time will decrease initially as we add more threads, but will then quickly level off (leftmost plot below). Although the maximum speedup of ~3.75x can be attained with 8-14 threads (middle plot), it corresponds to a rather poor parallel efficiency of 60%-30% (right plot). The recommended number of threads (4) yields a reasonable speedup (3x) without compromising the parallel efficiency too much (75%). Finally, if we use an excessively large number of cores (>=16 in this example), the execution time will start to increase again. Although the actual speedups will vary across datasets and systems, the general trend will stay the same. Therefore, it is up to the user to decide how much resources (=higher CPU time) can be sacrificed to obtain the results faster (=lower execution time).