infercnv tumor subclusters
By default, inferCNV operates at the level of whole samples, such as all cells defined as a certain cell type derived from a single patient. This is the fastest way to run inferCNV, but often not the optimal way, as a given tumor sample may have subpopulations with varied patterns of CNV.
By setting infercnv::run(analysis_mode='subclusters'), inferCNV will attempt to partition cells into groups having consistent patterns of CNV. CNV prediction (via HMM) would then be performed at the level of the subclusters rather than whole samples.
The view below shows differences obtained when performing HMM predictions at the level of whole samples as compared to subclusters.
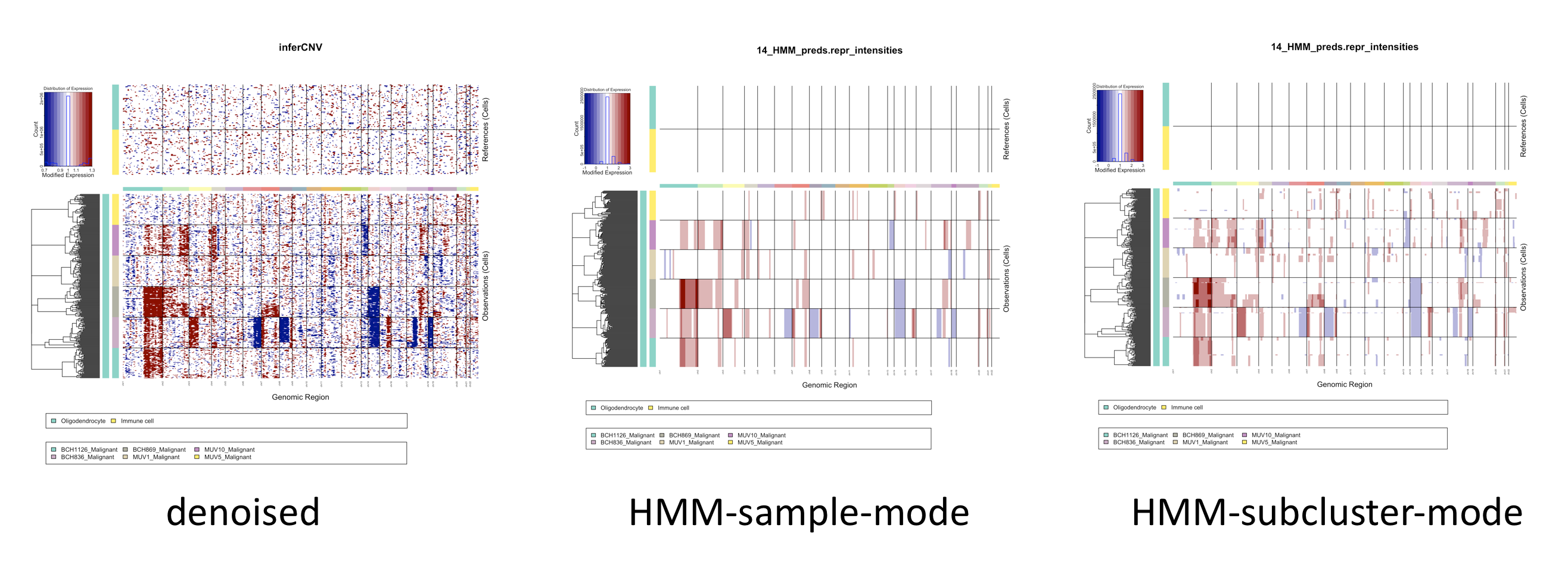
TODO: show version w/ more cells, gives better resolution for subclusters.
The methods available for defining tumor subclusters will continue to be expanded. We've currently had best success with using hierarchical clustering based methods.
There are 2 main categories of tumor subclustering available in infercnv: Leiden based and hclust based. The hclust based methods were the first to be added to infercnv and it is easier to see the effect of changing options as their core it to just run an hclust and then determine how many cuts to do in the resulting tree. Leiden based methods were later added for performance as the size of datasets increased because running an hclust (or multiple for random trees) scales poorly with the number of cells. Changes in options in the Leiden clustering are however harder to easily predict the effect of.
For running the Leiden clustering, the data to be subclustered can either used as is (at the end of step 14, so the data is the residual expression not denoised), or a PCA can be run on it to reduce dimensionality. A nearest neighbor search is then run to build a graph from the adjacency matrix, before being partioned according to the Leiden algorithm.
The parameters that impact the Leiden clustering based partitioning through infercnv::run() are:
- k_nn: Number k of nearest neighbors to search for when using the Leiden partition method for subclustering. (default: 20)
- leiden_method: Data preprocessing method used to generate the graph on which the Leiden algorithm is applied, one of "PCA" or "simple" (no extra preprocessing). (default: "PCA")
- leiden_function: Whether to use the Constant Potts Model (CPM) or modularity in igraph. Must be either "CPM" or "modularity". (default: "CPM")
- leiden_resolution: resolution parameter for the Leiden algorithm using the CPM quality score. For smaller datasets, 1 for modularity of 0.1 for CPM are a good start, but they usually need to be lowered as the size and diversity of the dataset increases. (default: auto)
There is a further set of options that allow to run an additional subclustering on each chromosome separately for the purposes of running the HMM alone. When used, each chromosome is processed by the HMM with its own set of independent subclusters to have more cells per subcluster. Then a consensus of the results across all cells of a given subclusters defined on all the data is taken for each subcluster. Because there are significantly fewer genes when running on a per chromosome basis, the default settings used are different for this mode.
- leiden_method_per_chr: Method used to generate the graph on which the Leiden algorithm is applied for the per chromosome subclustering, one of "PCA" or "simple". (default: "simple")
- leiden_function_per_chr: Whether to use the Constant Potts Model (CPM) or modularity in igraph for the per chromosome subclustering. Must be either "CPM" or "modularity". (default: "modularity")
- leiden_resolution_per_chr: resolution parameter for the Leiden algorithm for the per chromosome subclustering (default: 1)
- per_chr_hmm_subclusters: Run subclustering per chromosome over all cells combined to run the HMM on those subclusters instead. Only applicable when using Leiden subclustering. This should provide enough definition in the predictions while avoiding subclusters that are too small thus providing less evidence to work with. (default: FALSE)
- per_chr_hmm_subclusters_references: Whether the per chromosome subclustering should also be done on references, which should not have as much variation as observations. (default = FALSE)
The parameters that impact the hierarchical clustering based tree partitioning include:
-
'infercnv::run(hclust_method='ward.D2') : the clustering method to use. All built-in R hclust methods are supported: ("ward.D", "ward.D2", "single", "complete", "average", "mcquitty", "median", "centroid"). We find 'ward.D2' (default) to work best.
-
'infercnv::run(tumor_subcluster_partition_method='random_trees') : method used for partitioning the hierarchical clustering tree. Options include ('random_trees', 'qnorm'). These are described further below. Both methods rely on the 'infercnv::run(tumor_subcluster_pval=0.05)' setting for determining cut-points in the hierarchical tree.
This method was inspired by the SHC method. We utilize a non-parameteric method that involves comparing the hierarchical tree height to a null distribution of tree heights derived from trees involving randomly permuted genes. If the observed tree height is found to be statistically significant according to the 'infercnv::run(tumor_subcluster_pval=0.05)' setting, the tree is bifurcated. This procedure is then applied recursively to the split trees and splitting will continue to occur until a maximum recursion depth is reached, the clade under study has too few members, or the subtree height is found to not be significant under the corresponding null distribution.
An advantage of this method is that it will not partition a sample of cells if there's insufficient evidence for tumor heterogeneity. A disadvantage is that the method is relatively slow, given that it needs to perform 100 separate tree constructions at each tested bifurcation in order to generate a null distribution. However, parallelization is enabled and the 'infercnv::run(num_threads=4)' can be further increased to speed up the process.
This involves a parametric approach that cuts the hierarchical tree at the tree height quantile corresponding to the quantile of a normal distribution of the tree heights where the percentile = 'infercnv::run(tumor_subcluster_pval=0.05)'.
The advantage of this approach is that it is a fast approach for exploring groups of cells that may represent tumor heterogeneity instead of being restricted to running all cells through as a single sample. The disadvantage is that it will split the hierarchical tree even when there is no true statistical evidence for heterogeneity. It is really only a simple dynamic way of exploring potential heterogeneity during an inferCNV run.